 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTIDE: Trajectory-based Diagnostic Evaluation of Test-Time Improvement in LLM Agents
Feb 03, 2026Recent advances in autonomous LLM agents demonstrate their ability to improve performance through iterative interaction with the environment. We define this paradigm as Test-Time Improvement (TTI). However, the mechanisms under how and why TTI succeed or fail remain poorly understood, and existing evaluation metrics fail to capture their task optimization efficiency, behavior adaptation after erroneous actions, and the specific utility of working memory for task completion. To address these gaps, we propose Test-time Improvement Diagnostic Evaluation (TIDE), an agent-agnostic and environment-agnostic framework that decomposes TTI into three comprehensive and interconnected dimensions. The framework measures (1) the overall temporal dynamics of task completion and (2) identifies whether performance is primarily constrained by recursive looping behaviors or (3) by burdensome accumulated memory. Through extensive experiments across diverse agents and environments, TIDE highlights that improving agent performance requires more than scaling internal reasoning, calling for explicitly optimizing the interaction dynamics between the agent and the environment.
Scalable Machine Learning Force Fields for Macromolecular Systems Through Long-Range Aware Message Passing
Jan 07, 2026Machine learning force fields (MLFFs) have revolutionized molecular simulations by providing quantum mechanical accuracy at the speed of molecular mechanical computations. However, a fundamental reliance of these models on fixed-cutoff architectures limits their applicability to macromolecular systems where long-range interactions dominate. We demonstrate that this locality constraint causes force prediction errors to scale monotonically with system size, revealing a critical architectural bottleneck. To overcome this, we establish the systematically designed MolLR25 ({Mol}ecules with {L}ong-{R}ange effect) benchmark up to 1200 atoms, generated using high-fidelity DFT, and introduce E2Former-LSR, an equivariant transformer that explicitly integrates long-range attention blocks. E2Former-LSR exhibits stable error scaling, achieves superior fidelity in capturing non-covalent decay, and maintains precision on complex protein conformations. Crucially, its efficient design provides up to 30% speedup compared to purely local models. This work validates the necessity of non-local architectures for generalizable MLFFs, enabling high-fidelity molecular dynamics for large-scale chemical and biological systems.
MolChord: Structure-Sequence Alignment for Protein-Guided Drug Design
Oct 31, 2025Structure-based drug design (SBDD), which maps target proteins to candidate molecular ligands, is a fundamental task in drug discovery. Effectively aligning protein structural representations with molecular representations, and ensuring alignment between generated drugs and their pharmacological properties, remains a critical challenge. To address these challenges, we propose MolChord, which integrates two key techniques: (1) to align protein and molecule structures with their textual descriptions and sequential representations (e.g., FASTA for proteins and SMILES for molecules), we leverage NatureLM, an autoregressive model unifying text, small molecules, and proteins, as the molecule generator, alongside a diffusion-based structure encoder; and (2) to guide molecules toward desired properties, we curate a property-aware dataset by integrating preference data and refine the alignment process using Direct Preference Optimization (DPO). Experimental results on CrossDocked2020 demonstrate that our approach achieves state-of-the-art performance on key evaluation metrics, highlighting its potential as a practical tool for SBDD.
Instructing Text-to-Image Diffusion Models via Classifier-Guided Semantic Optimization
May 20, 2025Text-to-image diffusion models have emerged as powerful tools for high-quality image generation and editing. Many existing approaches rely on text prompts as editing guidance. However, these methods are constrained by the need for manual prompt crafting, which can be time-consuming, introduce irrelevant details, and significantly limit editing performance. In this work, we propose optimizing semantic embeddings guided by attribute classifiers to steer text-to-image models toward desired edits, without relying on text prompts or requiring any training or fine-tuning of the diffusion model. We utilize classifiers to learn precise semantic embeddings at the dataset level. The learned embeddings are theoretically justified as the optimal representation of attribute semantics, enabling disentangled and accurate edits. Experiments further demonstrate that our method achieves high levels of disentanglement and strong generalization across different domains of data.
Chain-of-Model Learning for Language Model
May 17, 2025In this paper, we propose a novel learning paradigm, termed Chain-of-Model (CoM), which incorporates the causal relationship into the hidden states of each layer as a chain style, thereby introducing great scaling efficiency in model training and inference flexibility in deployment. We introduce the concept of Chain-of-Representation (CoR), which formulates the hidden states at each layer as a combination of multiple sub-representations (i.e., chains) at the hidden dimension level. In each layer, each chain from the output representations can only view all of its preceding chains in the input representations. Consequently, the model built upon CoM framework can progressively scale up the model size by increasing the chains based on the previous models (i.e., chains), and offer multiple sub-models at varying sizes for elastic inference by using different chain numbers. Based on this principle, we devise Chain-of-Language-Model (CoLM), which incorporates the idea of CoM into each layer of Transformer architecture. Based on CoLM, we further introduce CoLM-Air by introducing a KV sharing mechanism, that computes all keys and values within the first chain and then shares across all chains. This design demonstrates additional extensibility, such as enabling seamless LM switching, prefilling acceleration and so on. Experimental results demonstrate our CoLM family can achieve comparable performance to the standard Transformer, while simultaneously enabling greater flexiblity, such as progressive scaling to improve training efficiency and offer multiple varying model sizes for elastic inference, paving a a new way toward building language models. Our code will be released in the future at: https://github.com/microsoft/CoLM.
MoonCast: High-Quality Zero-Shot Podcast Generation
Mar 19, 2025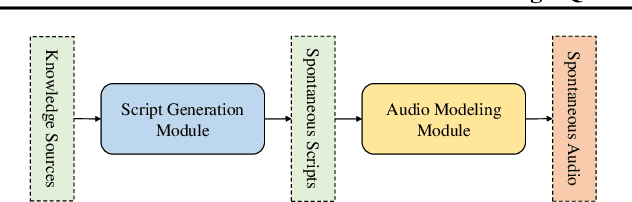


Recent advances in text-to-speech synthesis have achieved notable success in generating high-quality short utterances for individual speakers. However, these systems still face challenges when extending their capabilities to long, multi-speaker, and spontaneous dialogues, typical of real-world scenarios such as podcasts. These limitations arise from two primary challenges: 1) long speech: podcasts typically span several minutes, exceeding the upper limit of most existing work; 2) spontaneity: podcasts are marked by their spontaneous, oral nature, which sharply contrasts with formal, written contexts; existing works often fall short in capturing this spontaneity. In this paper, we propose MoonCast, a solution for high-quality zero-shot podcast generation, aiming to synthesize natural podcast-style speech from text-only sources (e.g., stories, technical reports, news in TXT, PDF, or Web URL formats) using the voices of unseen speakers. To generate long audio, we adopt a long-context language model-based audio modeling approach utilizing large-scale long-context speech data. To enhance spontaneity, we utilize a podcast generation module to generate scripts with spontaneous details, which have been empirically shown to be as crucial as the text-to-speech modeling itself. Experiments demonstrate that MoonCast outperforms baselines, with particularly notable improvements in spontaneity and coherence.
UniGenX: Unified Generation of Sequence and Structure with Autoregressive Diffusion
Mar 09, 2025Unified generation of sequence and structure for scientific data (e.g., materials, molecules, proteins) is a critical task. Existing approaches primarily rely on either autoregressive sequence models or diffusion models, each offering distinct advantages and facing notable limitations. Autoregressive models, such as GPT, Llama, and Phi-4, have demonstrated remarkable success in natural language generation and have been extended to multimodal tasks (e.g., image, video, and audio) using advanced encoders like VQ-VAE to represent complex modalities as discrete sequences. However, their direct application to scientific domains is challenging due to the high precision requirements and the diverse nature of scientific data. On the other hand, diffusion models excel at generating high-dimensional scientific data, such as protein, molecule, and material structures, with remarkable accuracy. Yet, their inability to effectively model sequences limits their potential as general-purpose multimodal foundation models. To address these challenges, we propose UniGenX, a unified framework that combines autoregressive next-token prediction with conditional diffusion models. This integration leverages the strengths of autoregressive models to ease the training of conditional diffusion models, while diffusion-based generative heads enhance the precision of autoregressive predictions. We validate the effectiveness of UniGenX on material and small molecule generation tasks, achieving a significant leap in state-of-the-art performance for material crystal structure prediction and establishing new state-of-the-art results for small molecule structure prediction, de novo design, and conditional generation. Notably, UniGenX demonstrates significant improvements, especially in handling long sequences for complex structures, showcasing its efficacy as a versatile tool for scientific data generation.
Fast and Accurate Blind Flexible Docking
Feb 20, 2025Molecular docking that predicts the bound structures of small molecules (ligands) to their protein targets, plays a vital role in drug discovery. However, existing docking methods often face limitations: they either overlook crucial structural changes by assuming protein rigidity or suffer from low computational efficiency due to their reliance on generative models for structure sampling. To address these challenges, we propose FABFlex, a fast and accurate regression-based multi-task learning model designed for realistic blind flexible docking scenarios, where proteins exhibit flexibility and binding pocket sites are unknown (blind). Specifically, FABFlex's architecture comprises three specialized modules working in concert: (1) A pocket prediction module that identifies potential binding sites, addressing the challenges inherent in blind docking scenarios. (2) A ligand docking module that predicts the bound (holo) structures of ligands from their unbound (apo) states. (3) A pocket docking module that forecasts the holo structures of protein pockets from their apo conformations. Notably, FABFlex incorporates an iterative update mechanism that serves as a conduit between the ligand and pocket docking modules, enabling continuous structural refinements. This approach effectively integrates the three subtasks of blind flexible docking-pocket identification, ligand conformation prediction, and protein flexibility modeling-into a unified, coherent framework. Extensive experiments on public benchmark datasets demonstrate that FABFlex not only achieves superior effectiveness in predicting accurate binding modes but also exhibits a significant speed advantage (208 $\times$) compared to existing state-of-the-art methods. Our code is released at https://github.com/tmlr-group/FABFlex.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
\underline{E2}Former: A Linear-time \underline{E}fficient and \underline{E}quivariant Trans\underline{former} for Scalable Molecular Modeling
Jan 31, 2025



Equivariant Graph Neural Networks (EGNNs) have demonstrated significant success in modeling microscale systems, including those in chemistry, biology and materials science. However, EGNNs face substantial computational challenges due to the high cost of constructing edge features via spherical tensor products, making them impractical for large-scale systems. To address this limitation, we introduce E2Former, an equivariant and efficient transformer architecture that incorporates the Wigner $6j$ convolution (Wigner $6j$ Conv). By shifting the computational burden from edges to nodes, the Wigner $6j$ Conv reduces the complexity from $O(|\mathcal{E}|)$ to $ O(| \mathcal{V}|)$ while preserving both the model's expressive power and rotational equivariance. We show that this approach achieves a 7x-30x speedup compared to conventional $\mathrm{SO}(3)$ convolutions. Furthermore, our empirical results demonstrate that the derived E2Former mitigates the computational challenges of existing approaches without compromising the ability to capture detailed geometric information. This development could suggest a promising direction for scalable and efficient molecular modeling.