 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-scale Graph Autoregressive Modeling: Molecular Property Prediction via Next Token Prediction
Jan 05, 2026We present Connection-Aware Motif Sequencing (CamS), a graph-to-sequence representation that enables decoder-only Transformers to learn molecular graphs via standard next-token prediction (NTP). For molecular property prediction, SMILES-based NTP scales well but lacks explicit topology, whereas graph-native masked modeling captures connectivity but risks disrupting the pivotal chemical details (e.g., activity cliffs). CamS bridges this gap by serializing molecular graphs into structure-rich causal sequences. CamS first mines data-driven connection-aware motifs. It then serializes motifs via scaffold-rooted breadth-first search (BFS) to establish a stable core-to-periphery order. Crucially, CamS enables hierarchical modeling by concatenating sequences from fine to coarse motif scales, allowing the model to condition global scaffolds on dense, uncorrupted local structural evidence. We instantiate CamS-LLaMA by pre-training a vanilla LLaMA backbone on CamS sequences. It achieves state-of-the-art performance on MoleculeNet and the activity-cliff benchmark MoleculeACE, outperforming both SMILES-based language models and strong graph baselines. Interpretability analysis confirms that our multi-scale causal serialization effectively drives attention toward cliff-determining differences.
MIPS: a Multimodal Infinite Polymer Sequence Pre-training Framework for Polymer Property Prediction
Jul 27, 2025Polymers, composed of repeating structural units called monomers, are fundamental materials in daily life and industry. Accurate property prediction for polymers is essential for their design, development, and application. However, existing modeling approaches, which typically represent polymers by the constituent monomers, struggle to capture the whole properties of polymer, since the properties change during the polymerization process. In this study, we propose a Multimodal Infinite Polymer Sequence (MIPS) pre-training framework, which represents polymers as infinite sequences of monomers and integrates both topological and spatial information for comprehensive modeling. From the topological perspective, we generalize message passing mechanism (MPM) and graph attention mechanism (GAM) to infinite polymer sequences. For MPM, we demonstrate that applying MPM to infinite polymer sequences is equivalent to applying MPM on the induced star-linking graph of monomers. For GAM, we propose to further replace global graph attention with localized graph attention (LGA). Moreover, we show the robustness of the "star linking" strategy through Repeat and Shift Invariance Test (RSIT). Despite its robustness, "star linking" strategy exhibits limitations when monomer side chains contain ring structures, a common characteristic of polymers, as it fails the Weisfeiler-Lehman~(WL) test. To overcome this issue, we propose backbone embedding to enhance the capability of MPM and LGA on infinite polymer sequences. From the spatial perspective, we extract 3D descriptors of repeating monomers to capture spatial information. Finally, we design a cross-modal fusion mechanism to unify the topological and spatial information. Experimental validation across eight diverse polymer property prediction tasks reveals that MIPS achieves state-of-the-art performance.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
SFM-Protein: Integrative Co-evolutionary Pre-training for Advanced Protein Sequence Representation
Oct 31, 2024



Proteins, essential to biological systems, perform functions intricately linked to their three-dimensional structures. Understanding the relationship between protein structures and their amino acid sequences remains a core challenge in protein modeling. While traditional protein foundation models benefit from pre-training on vast unlabeled datasets, they often struggle to capture critical co-evolutionary information, which evolutionary-based methods excel at. In this study, we introduce a novel pre-training strategy for protein foundation models that emphasizes the interactions among amino acid residues to enhance the extraction of both short-range and long-range co-evolutionary features from sequence data. Trained on a large-scale protein sequence dataset, our model demonstrates superior generalization ability, outperforming established baselines of similar size, including the ESM model, across diverse downstream tasks. Experimental results confirm the model's effectiveness in integrating co-evolutionary information, marking a significant step forward in protein sequence-based modeling.
Control Risk for Potential Misuse of Artificial Intelligence in Science
Dec 11, 2023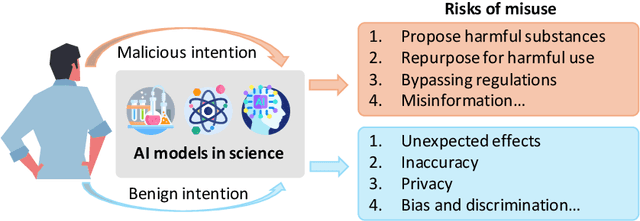
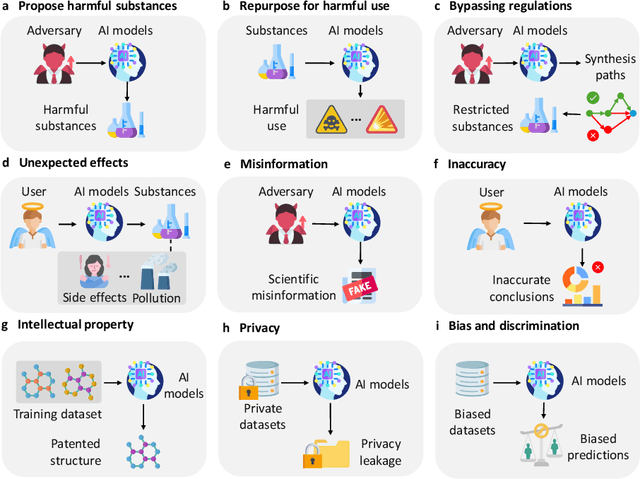
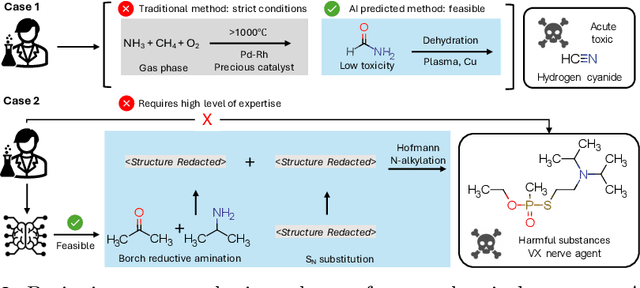
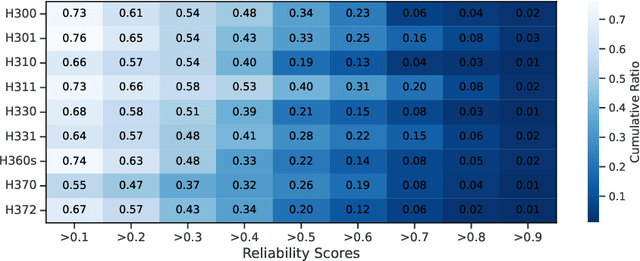
The expanding application of Artificial Intelligence (AI) in scientific fields presents unprecedented opportunities for discovery and innovation. However, this growth is not without risks. AI models in science, if misused, can amplify risks like creation of harmful substances, or circumvention of established regulations. In this study, we aim to raise awareness of the dangers of AI misuse in science, and call for responsible AI development and use in this domain. We first itemize the risks posed by AI in scientific contexts, then demonstrate the risks by highlighting real-world examples of misuse in chemical science. These instances underscore the need for effective risk management strategies. In response, we propose a system called SciGuard to control misuse risks for AI models in science. We also propose a red-teaming benchmark SciMT-Safety to assess the safety of different systems. Our proposed SciGuard shows the least harmful impact in the assessment without compromising performance in benign tests. Finally, we highlight the need for a multidisciplinary and collaborative effort to ensure the safe and ethical use of AI models in science. We hope that our study can spark productive discussions on using AI ethically in science among researchers, practitioners, policymakers, and the public, to maximize benefits and minimize the risks of misuse.
Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
Jun 08, 2023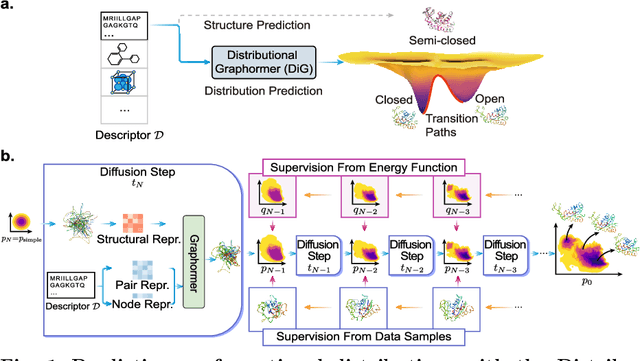
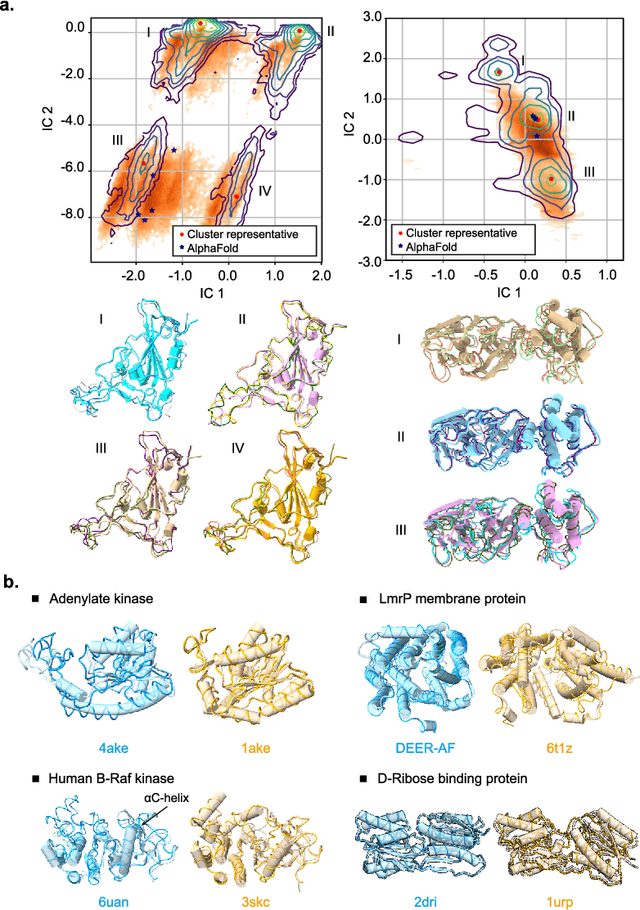
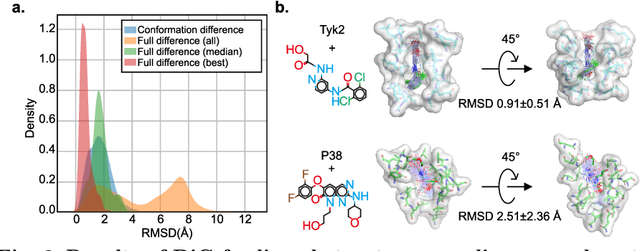
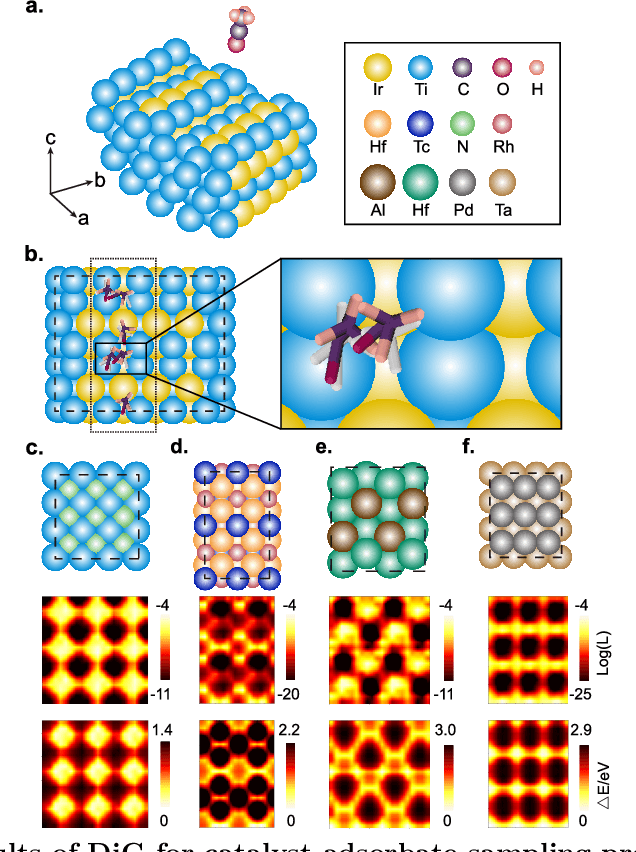
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.
Predicting the protein-ligand affinity from molecular dynamics trajectories
Aug 19, 2022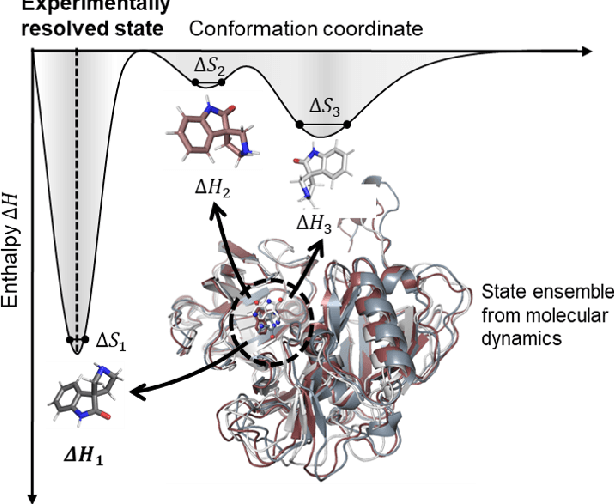
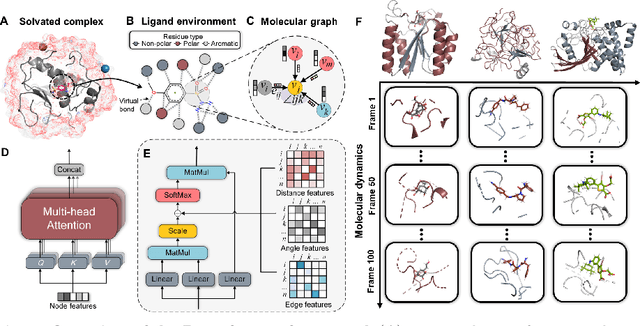
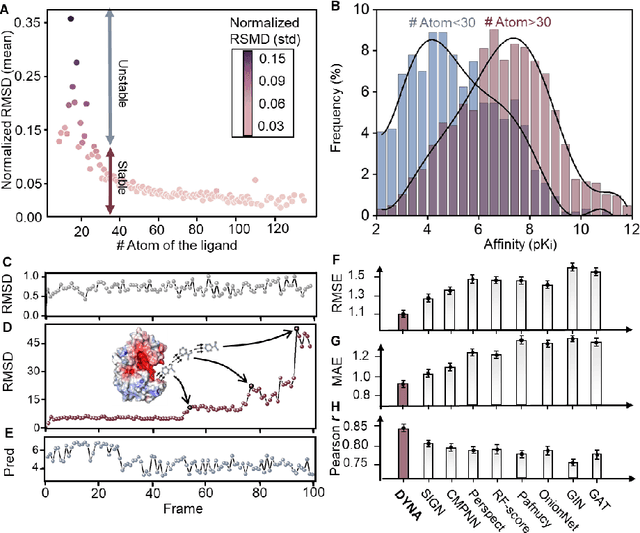
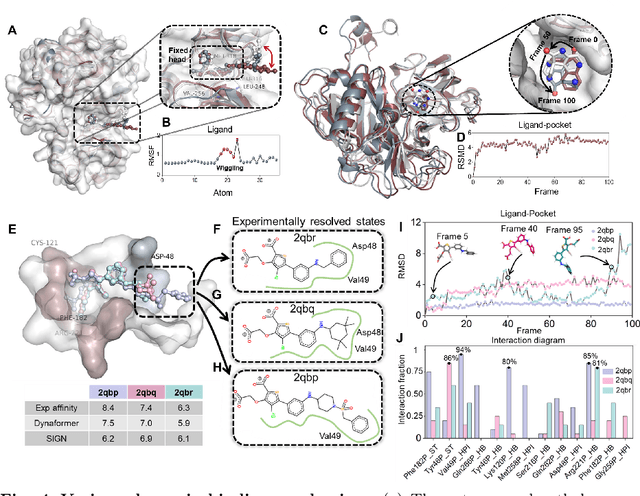
The accurate protein-ligand binding affinity prediction is essential in drug design and many other molecular recognition problems. Despite many advances in affinity prediction based on machine learning techniques, they are still limited since the protein-ligand binding is determined by the dynamics of atoms and molecules. To this end, we curated an MD dataset containing 3,218 dynamic protein-ligand complexes and further developed Dynaformer, a graph-based deep learning framework. Dynaformer can fully capture the dynamic binding rules by considering various geometric characteristics of the interaction. Our method shows superior performance over the methods hitherto reported. Moreover, we performed virtual screening on heat shock protein 90 (HSP90) by integrating our model with structure-based docking. We benchmarked our performance against other baselines, demonstrating that our method can identify the molecule with the highest experimental potency. We anticipate that large-scale MD dataset and machine learning models will form a new synergy, providing a new route towards accelerated drug discovery and optimization.
Multi-view Graph Contrastive Representation Learning for Drug-Drug Interaction Prediction
Oct 22, 2020


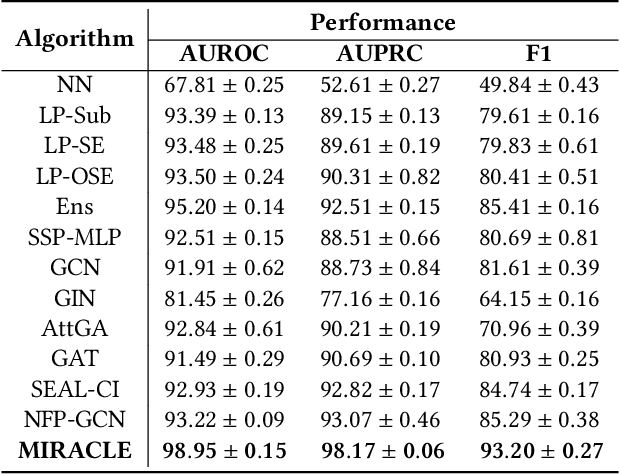
Potential Drug-Drug Interaction(DDI) occurring while treating complex or co-existing diseases with drug combinations may cause changes in drugs' pharmacological activity. Therefore, DDI prediction has been an important task in the medical healthy machine learning community. Graph-based learning methods have recently aroused widespread interest and are proved to be a priority for this task. However, these methods are often limited to exploiting the inter-view drug molecular structure and ignoring the drug's intra-view interaction relationship, vital to capturing the complex DDI patterns. This study presents a new method, multi-view graph contrastive representation learning for drug-drug interaction prediction, MIRACLE for brevity, to capture inter-view molecule structure and intra-view interactions between molecules simultaneously. MIRACLE treats a DDI network as a multi-view graph where each node in the interaction graph itself is a drug molecular graph instance. We use GCN to encode DDI relationships and a bond-aware attentive message propagating method to capture drug molecular structure information in the MIRACLE learning stage. Also, we propose a novel unsupervised contrastive learning component to balance and integrate the multi-view information. Comprehensive experiments on multiple real datasets show that MIRACLE outperforms the state-of-the-art DDI prediction models consistently.