 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Confidential Computing on nVIDIA H100 GPU: A Performance Benchmark Study
Sep 06, 2024This report evaluates the performance impact of enabling Trusted Execution Environments (TEE) on NVIDIA H100 GPUs for large language model (LLM) inference tasks. We benchmark the overhead introduced by TEE mode across various models and token lengths, focusing on the bottleneck caused by CPU-GPU data transfers via PCIe. Our results show that while there is minimal computational overhead within the GPU, the overall performance penalty is primarily due to data transfer. For most typical LLM queries, the overhead remains below 5%, with larger models and longer sequences experiencing near-zero overhead.
Ultrasound Nodule Segmentation Using Asymmetric Learning with Simple Clinical Annotation
Apr 23, 2024Recent advances in deep learning have greatly facilitated the automated segmentation of ultrasound images, which is essential for nodule morphological analysis. Nevertheless, most existing methods depend on extensive and precise annotations by domain experts, which are labor-intensive and time-consuming. In this study, we suggest using simple aspect ratio annotations directly from ultrasound clinical diagnoses for automated nodule segmentation. Especially, an asymmetric learning framework is developed by extending the aspect ratio annotations with two types of pseudo labels, i.e., conservative labels and radical labels, to train two asymmetric segmentation networks simultaneously. Subsequently, a conservative-radical-balance strategy (CRBS) strategy is proposed to complementally combine radical and conservative labels. An inconsistency-aware dynamically mixed pseudo-labels supervision (IDMPS) module is introduced to address the challenges of over-segmentation and under-segmentation caused by the two types of labels. To further leverage the spatial prior knowledge provided by clinical annotations, we also present a novel loss function namely the clinical anatomy prior loss. Extensive experiments on two clinically collected ultrasound datasets (thyroid and breast) demonstrate the superior performance of our proposed method, which can achieve comparable and even better performance than fully supervised methods using ground truth annotations.
Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
Jun 08, 2023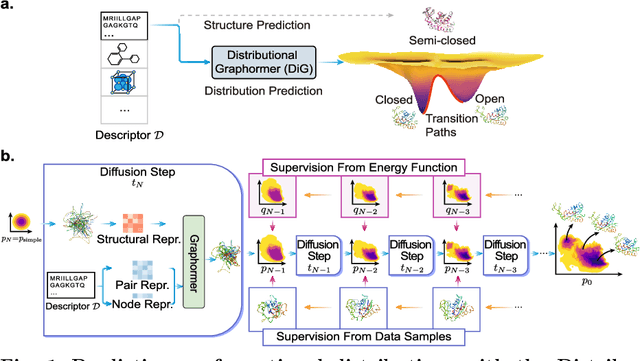
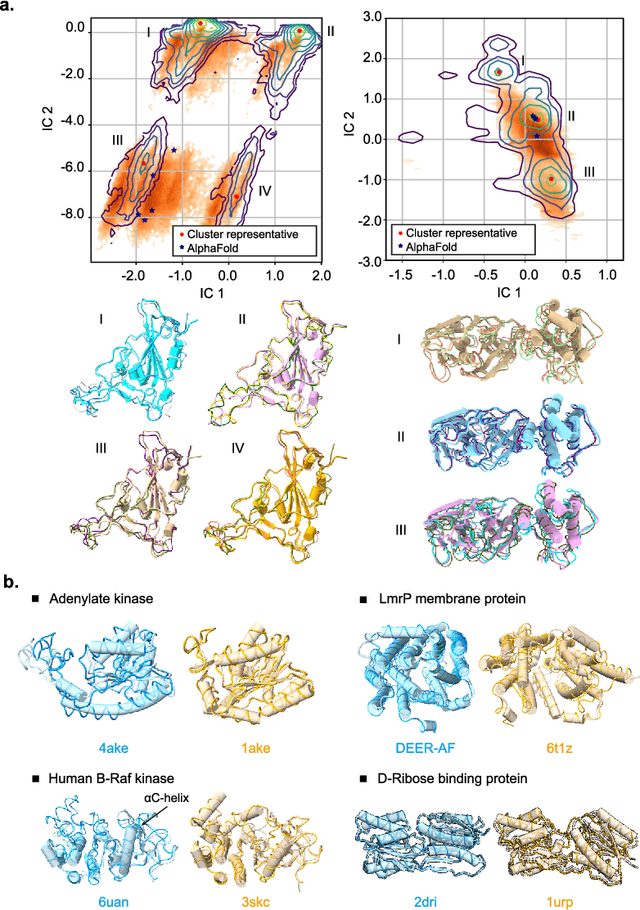
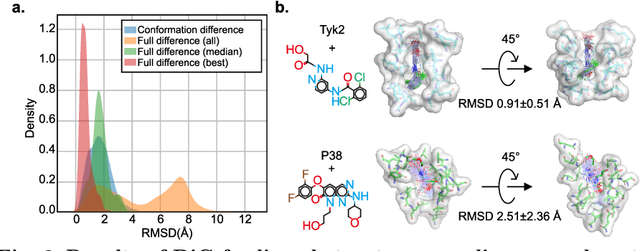
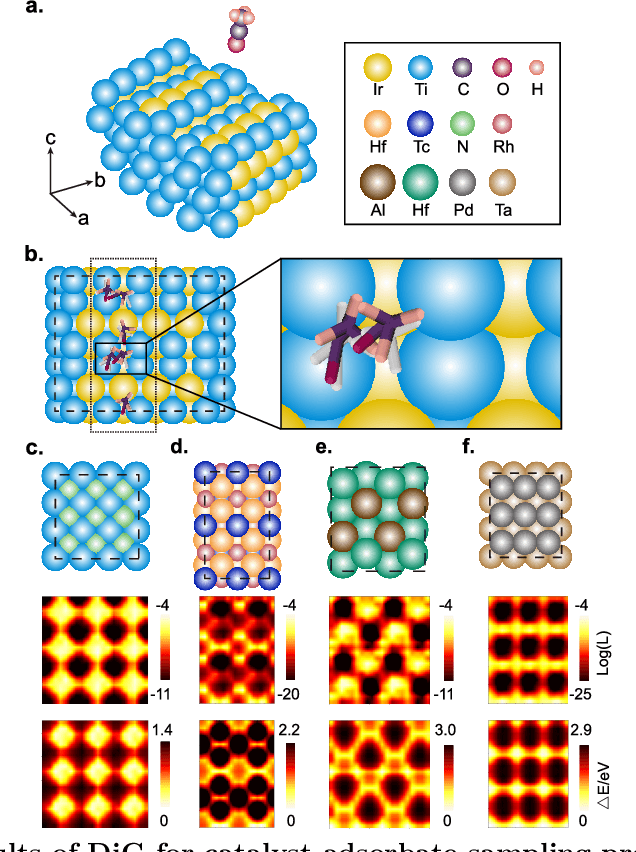
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.
Pre-training Co-evolutionary Protein Representation via A Pairwise Masked Language Model
Oct 29, 2021
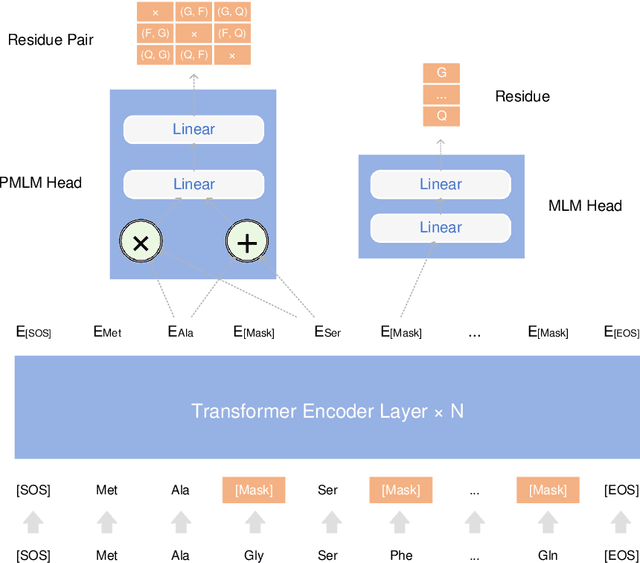
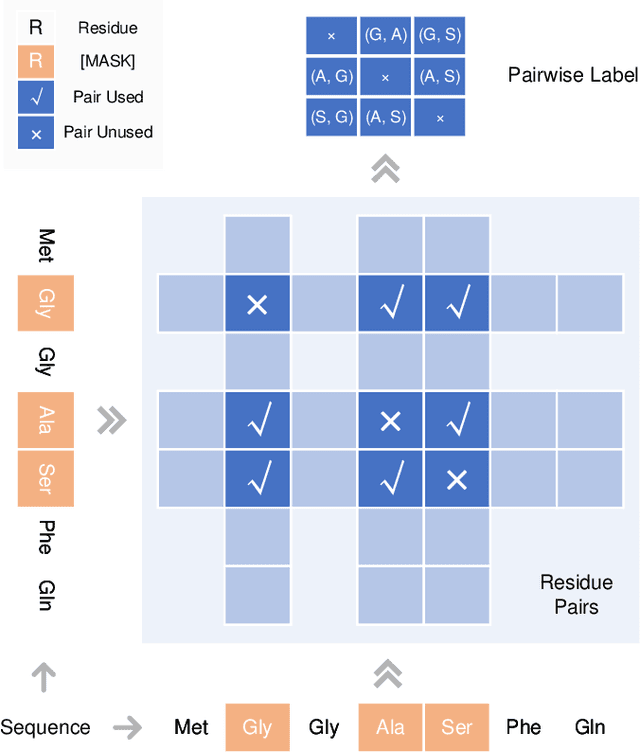
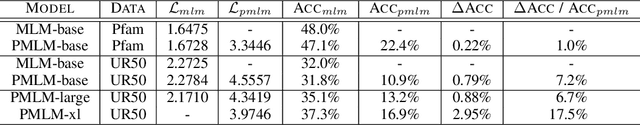
Understanding protein sequences is vital and urgent for biology, healthcare, and medicine. Labeling approaches are expensive yet time-consuming, while the amount of unlabeled data is increasing quite faster than that of the labeled data due to low-cost, high-throughput sequencing methods. In order to extract knowledge from these unlabeled data, representation learning is of significant value for protein-related tasks and has great potential for helping us learn more about protein functions and structures. The key problem in the protein sequence representation learning is to capture the co-evolutionary information reflected by the inter-residue co-variation in the sequences. Instead of leveraging multiple sequence alignment as is usually done, we propose a novel method to capture this information directly by pre-training via a dedicated language model, i.e., Pairwise Masked Language Model (PMLM). In a conventional masked language model, the masked tokens are modeled by conditioning on the unmasked tokens only, but processed independently to each other. However, our proposed PMLM takes the dependency among masked tokens into consideration, i.e., the probability of a token pair is not equal to the product of the probability of the two tokens. By applying this model, the pre-trained encoder is able to generate a better representation for protein sequences. Our result shows that the proposed method can effectively capture the inter-residue correlations and improves the performance of contact prediction by up to 9% compared to the MLM baseline under the same setting. The proposed model also significantly outperforms the MSA baseline by more than 7% on the TAPE contact prediction benchmark when pre-trained on a subset of the sequence database which the MSA is generated from, revealing the potential of the sequence pre-training method to surpass MSA based methods in general.
Seq-SetNet: Exploring Sequence Sets for Inferring Structures
Jun 06, 2019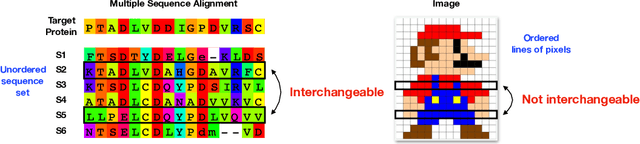
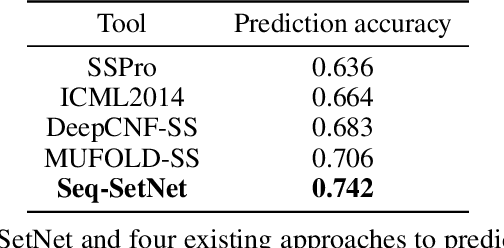
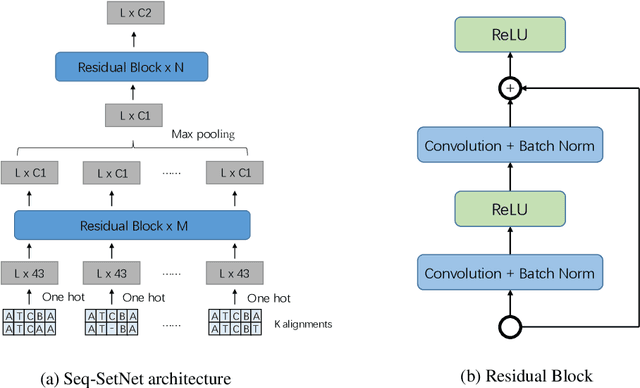
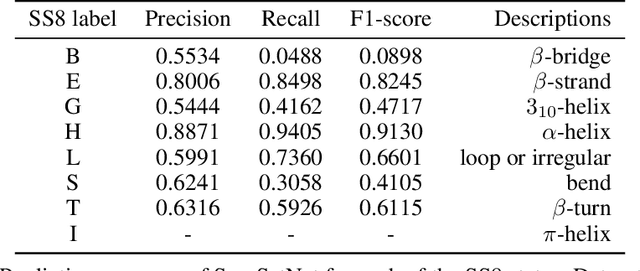
Sequence set is a widely-used type of data source in a large variety of fields. A typical example is protein structure prediction, which takes an multiple sequence alignment (MSA) as input and aims to infer structural information from it. Almost all of the existing approaches exploit MSAs in an indirect fashion, i.e., they transform MSAs into position-specific scoring matrices (PSSM) that represent the distribution of amino acid types at each column. PSSM could capture column-wise characteristics of MSA, however, the column-wise characteristics embedded in each individual component sequence were nearly totally neglected. The drawback of PSSM is rooted in the fact that an MSA is essentially an unordered sequence set rather than a matrix. Specifically, the interchange of any two sequences will not affect the whole MSA. In contrast, the pixels in an image essentially form a matrix since any two rows of pixels cannot be interchanged. Therefore, the traditional deep neural networks designed for image processing cannot be directly applied on sequence sets. Here, we proposed a novel deep neural network framework (called Seq-SetNet) for sequence set processing. By employing a {\it symmetric function} module to integrate features calculated from preceding layers, Seq-SetNet are immune to the order of sequences in the input MSA. This advantage enables us to directly and fully exploit MSAs by considering each component protein individually. We evaluated Seq-SetNet by using it to extract structural information from MSA for protein secondary structure prediction. Experimental results on popular benchmark sets suggests that Seq-SetNet outperforms the state-of-the-art approaches by 3.6% in precision. These results clearly suggest the advantages of Seq-SetNet in sequence set processing and it can be readily used in a wide range of fields, say natural language processing.
Predicting protein inter-residue contacts using composite likelihood maximization and deep learning
Aug 31, 2018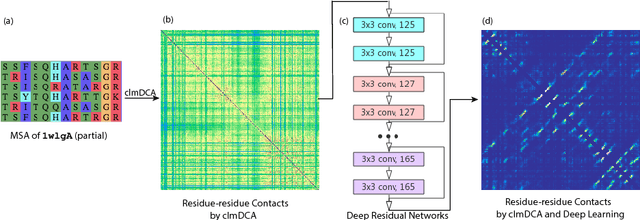
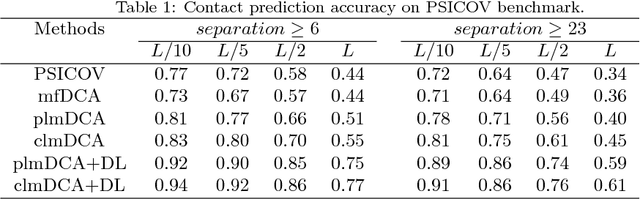
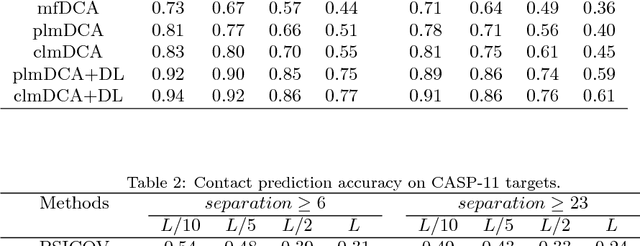
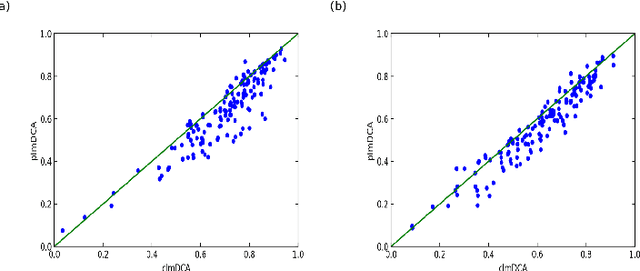
Accurate prediction of inter-residue contacts of a protein is important to calcu- lating its tertiary structure. Analysis of co-evolutionary events among residues has been proved effective to inferring inter-residue contacts. The Markov ran- dom field (MRF) technique, although being widely used for contact prediction, suffers from the following dilemma: the actual likelihood function of MRF is accurate but time-consuming to calculate, in contrast, approximations to the actual likelihood, say pseudo-likelihood, are efficient to calculate but inaccu- rate. Thus, how to achieve both accuracy and efficiency simultaneously remains a challenge. In this study, we present such an approach (called clmDCA) for contact prediction. Unlike plmDCA using pseudo-likelihood, i.e., the product of conditional probability of individual residues, our approach uses composite- likelihood, i.e., the product of conditional probability of all residue pairs. Com- posite likelihood has been theoretically proved as a better approximation to the actual likelihood function than pseudo-likelihood. Meanwhile, composite likelihood is still efficient to maximize, thus ensuring the efficiency of clmDCA. We present comprehensive experiments on popular benchmark datasets, includ- ing PSICOV dataset and CASP-11 dataset, to show that: i) clmDCA alone outperforms the existing MRF-based approaches in prediction accuracy. ii) When equipped with deep learning technique for refinement, the prediction ac- curacy of clmDCA was further significantly improved, suggesting the suitability of clmDCA for subsequent refinement procedure. We further present successful application of the predicted contacts to accurately build tertiary structures for proteins in the PSICOV dataset. Accessibility: The software clmDCA and a server are publicly accessible through http://protein.ict.ac.cn/clmDCA/.