 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproving Molecular Graph Generation with Flow Matching and Optimal Transport
Nov 08, 2024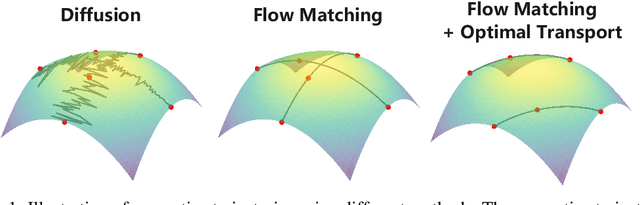


Generating molecular graphs is crucial in drug design and discovery but remains challenging due to the complex interdependencies between nodes and edges. While diffusion models have demonstrated their potentiality in molecular graph design, they often suffer from unstable training and inefficient sampling. To enhance generation performance and training stability, we propose GGFlow, a discrete flow matching generative model incorporating optimal transport for molecular graphs and it incorporates an edge-augmented graph transformer to enable the direct communications among chemical bounds. Additionally, GGFlow introduces a novel goal-guided generation framework to control the generative trajectory of our model, aiming to design novel molecular structures with the desired properties. GGFlow demonstrates superior performance on both unconditional and conditional molecule generation tasks, outperforming existing baselines and underscoring its effectiveness and potential for wider application.
The learned range test method for the inverse inclusion problem
Nov 01, 2024We consider the inverse problem consisting of the reconstruction of an inclusion $B$ contained in a bounded domain $\Omega\subset\mathbb{R}^d$ from a single pair of Cauchy data $(u|_{\partial\Omega},\partial_\nu u|_{\partial\Omega})$, where $\Delta u=0$ in $\Omega\setminus\overline B$ and $u=0$ on $\partial B$. We show that the reconstruction algorithm based on the range test, a domain sampling method, can be written as a neural network with a specific architecture. We propose to learn the weights of this network in the framework of supervised learning, and to combine it with a pre-trained classifier, with the purpose of distinguishing the inclusions based on their distance from the boundary. The numerical simulations show that this learned range test method provides accurate and stable reconstructions of polygonal inclusions. Furthermore, the results are superior to those obtained with the standard range test method (without learning) and with an end-to-end fully connected deep neural network, a purely data-driven method.
Seq-SetNet: Exploring Sequence Sets for Inferring Structures
Jun 06, 2019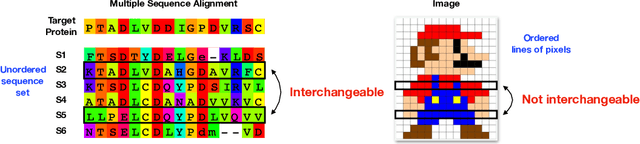
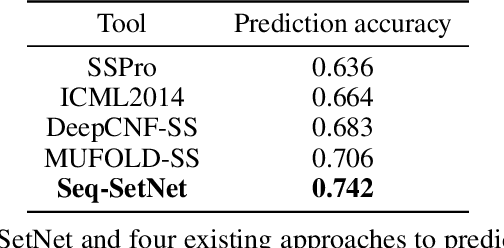
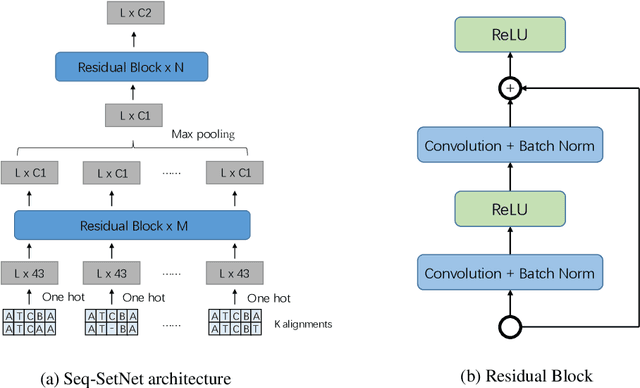
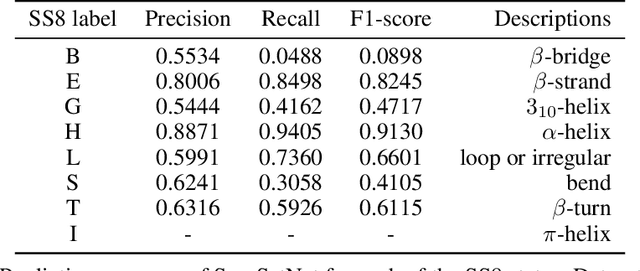
Sequence set is a widely-used type of data source in a large variety of fields. A typical example is protein structure prediction, which takes an multiple sequence alignment (MSA) as input and aims to infer structural information from it. Almost all of the existing approaches exploit MSAs in an indirect fashion, i.e., they transform MSAs into position-specific scoring matrices (PSSM) that represent the distribution of amino acid types at each column. PSSM could capture column-wise characteristics of MSA, however, the column-wise characteristics embedded in each individual component sequence were nearly totally neglected. The drawback of PSSM is rooted in the fact that an MSA is essentially an unordered sequence set rather than a matrix. Specifically, the interchange of any two sequences will not affect the whole MSA. In contrast, the pixels in an image essentially form a matrix since any two rows of pixels cannot be interchanged. Therefore, the traditional deep neural networks designed for image processing cannot be directly applied on sequence sets. Here, we proposed a novel deep neural network framework (called Seq-SetNet) for sequence set processing. By employing a {\it symmetric function} module to integrate features calculated from preceding layers, Seq-SetNet are immune to the order of sequences in the input MSA. This advantage enables us to directly and fully exploit MSAs by considering each component protein individually. We evaluated Seq-SetNet by using it to extract structural information from MSA for protein secondary structure prediction. Experimental results on popular benchmark sets suggests that Seq-SetNet outperforms the state-of-the-art approaches by 3.6% in precision. These results clearly suggest the advantages of Seq-SetNet in sequence set processing and it can be readily used in a wide range of fields, say natural language processing.
Predicting protein inter-residue contacts using composite likelihood maximization and deep learning
Aug 31, 2018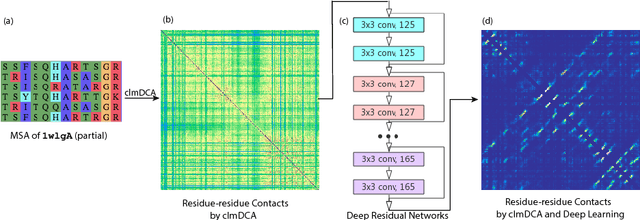
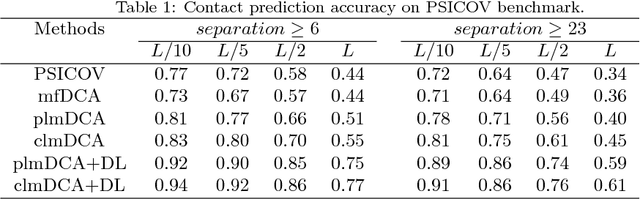
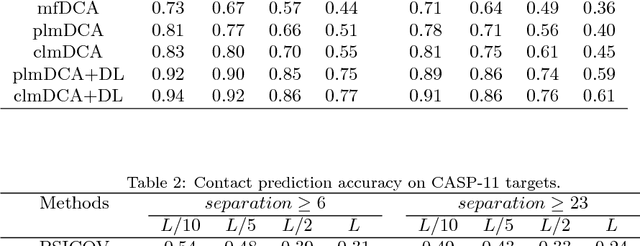
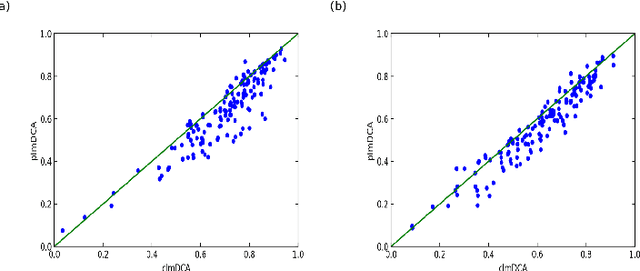
Accurate prediction of inter-residue contacts of a protein is important to calcu- lating its tertiary structure. Analysis of co-evolutionary events among residues has been proved effective to inferring inter-residue contacts. The Markov ran- dom field (MRF) technique, although being widely used for contact prediction, suffers from the following dilemma: the actual likelihood function of MRF is accurate but time-consuming to calculate, in contrast, approximations to the actual likelihood, say pseudo-likelihood, are efficient to calculate but inaccu- rate. Thus, how to achieve both accuracy and efficiency simultaneously remains a challenge. In this study, we present such an approach (called clmDCA) for contact prediction. Unlike plmDCA using pseudo-likelihood, i.e., the product of conditional probability of individual residues, our approach uses composite- likelihood, i.e., the product of conditional probability of all residue pairs. Com- posite likelihood has been theoretically proved as a better approximation to the actual likelihood function than pseudo-likelihood. Meanwhile, composite likelihood is still efficient to maximize, thus ensuring the efficiency of clmDCA. We present comprehensive experiments on popular benchmark datasets, includ- ing PSICOV dataset and CASP-11 dataset, to show that: i) clmDCA alone outperforms the existing MRF-based approaches in prediction accuracy. ii) When equipped with deep learning technique for refinement, the prediction ac- curacy of clmDCA was further significantly improved, suggesting the suitability of clmDCA for subsequent refinement procedure. We further present successful application of the predicted contacts to accurately build tertiary structures for proteins in the PSICOV dataset. Accessibility: The software clmDCA and a server are publicly accessible through http://protein.ict.ac.cn/clmDCA/.