 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDesigning faster mixed integer linear programming algorithm via learning the optimal path
Jan 22, 2026Designing faster algorithms for solving Mixed-Integer Linear Programming (MILP) problems is highly desired across numerous practical domains, as a vast array of complex real-world challenges can be effectively modeled as MILP formulations. Solving these problems typically employs the branch-and-bound algorithm, the core of which can be conceived as searching for a path of nodes (or sub-problems) that contains the optimal solution to the original MILP problem. Traditional approaches to finding this path rely heavily on hand-crafted, intuition-based heuristic strategies, which often suffer from unstable and unpredictable performance across different MILP problem instances. To address this limitation, we introduce DeepBound, a deep learning-based node selection algorithm that automates the learning of such human intuition from data. The core of DeepBound lies in learning to prioritize nodes containing the optimal solution, thereby improving solving efficiency. DeepBound introduces a multi-level feature fusion network to capture the node representations. To tackle the inherent node imbalance in branch-and-bound trees, DeepBound employs a pairwise training paradigm that enhances the model's ability to discriminate between nodes. Extensive experiments on three NP-hard MILP benchmarks demonstrate that DeepBound achieves superior solving efficiency over conventional heuristic rules and existing learning-based approaches, obtaining optimal feasible solutions with significantly reduced computation time. Moreover, DeepBound demonstrates strong generalization capability on large and complex instances. The analysis of its learned features reveals that the method can automatically discover more flexible and robust feature selection, which may effectively improve and potentially replace human-designed heuristic rules.
Improving Molecular Graph Generation with Flow Matching and Optimal Transport
Nov 08, 2024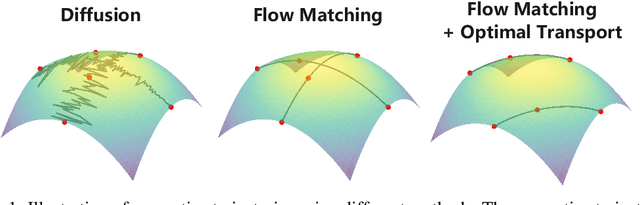

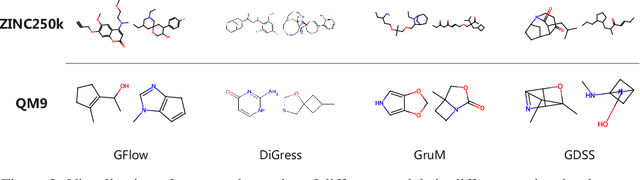

Generating molecular graphs is crucial in drug design and discovery but remains challenging due to the complex interdependencies between nodes and edges. While diffusion models have demonstrated their potentiality in molecular graph design, they often suffer from unstable training and inefficient sampling. To enhance generation performance and training stability, we propose GGFlow, a discrete flow matching generative model incorporating optimal transport for molecular graphs and it incorporates an edge-augmented graph transformer to enable the direct communications among chemical bounds. Additionally, GGFlow introduces a novel goal-guided generation framework to control the generative trajectory of our model, aiming to design novel molecular structures with the desired properties. GGFlow demonstrates superior performance on both unconditional and conditional molecule generation tasks, outperforming existing baselines and underscoring its effectiveness and potential for wider application.
Understanding is Compression
Jun 24, 2024We have previously shown all understanding or learning are compression, under reasonable assumptions. In principle, better understanding of data should improve data compression. Traditional compression methodologies focus on encoding frequencies or some other computable properties of data. Large language models approximate the uncomputable Solomonoff distribution, opening up a whole new avenue to justify our theory. Under the new uncomputable paradigm, we present LMCompress based on the understanding of data using large models. LMCompress has significantly better lossless compression ratios than all other lossless data compression methods, doubling the compression ratios of JPEG-XL for images, FLAC for audios and H264 for videos, and tripling or quadrupling the compression ratio of bz2 for texts. The better a large model understands the data, the better LMCompress compresses.
Predicting mutational effects on protein-protein binding via a side-chain diffusion probabilistic model
Oct 30, 2023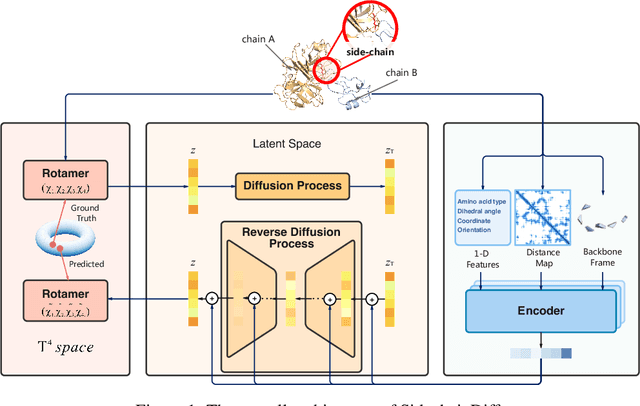
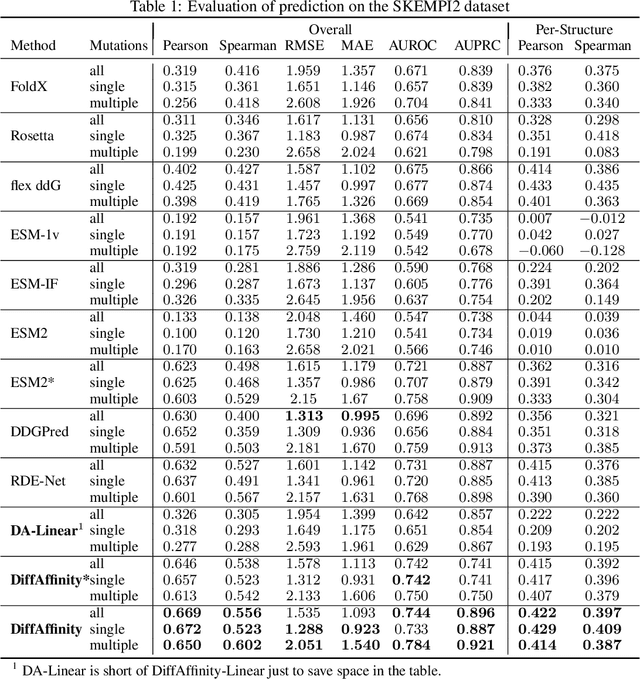
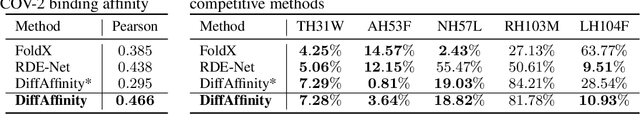
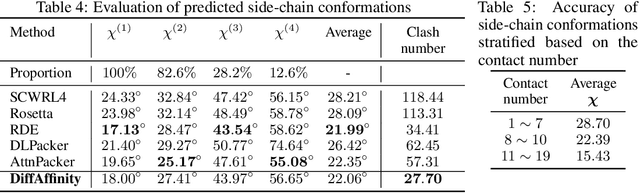
Many crucial biological processes rely on networks of protein-protein interactions. Predicting the effect of amino acid mutations on protein-protein binding is vital in protein engineering and therapeutic discovery. However, the scarcity of annotated experimental data on binding energy poses a significant challenge for developing computational approaches, particularly deep learning-based methods. In this work, we propose SidechainDiff, a representation learning-based approach that leverages unlabelled experimental protein structures. SidechainDiff utilizes a Riemannian diffusion model to learn the generative process of side-chain conformations and can also give the structural context representations of mutations on the protein-protein interface. Leveraging the learned representations, we achieve state-of-the-art performance in predicting the mutational effects on protein-protein binding. Furthermore, SidechainDiff is the first diffusion-based generative model for side-chains, distinguishing it from prior efforts that have predominantly focused on generating protein backbone structures.
A Theory of Human-Like Few-Shot Learning
Jan 03, 2023



We aim to bridge the gap between our common-sense few-sample human learning and large-data machine learning. We derive a theory of human-like few-shot learning from von-Neuman-Landauer's principle. modelling human learning is difficult as how people learn varies from one to another. Under commonly accepted definitions, we prove that all human or animal few-shot learning, and major models including Free Energy Principle and Bayesian Program Learning that model such learning, approximate our theory, under Church-Turing thesis. We find that deep generative model like variational autoencoder (VAE) can be used to approximate our theory and perform significantly better than baseline models including deep neural networks, for image recognition, low resource language processing, and character recognition.
NIERT: Accurate Numerical Interpolation through Unifying Scattered Data Representations using Transformer Encoder
Oct 07, 2022

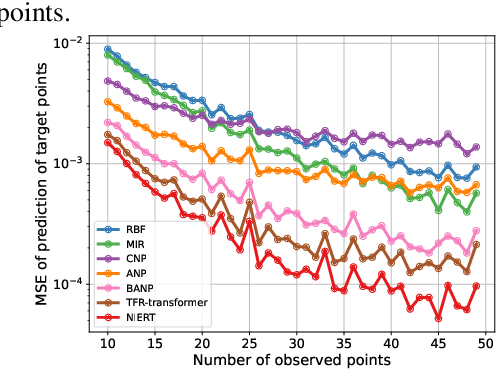
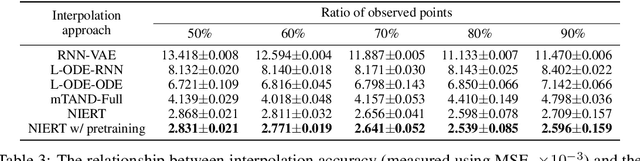
Numerical interpolation for scattered data, i.e., estimating values for target points based on those of some observed points, is widely used in computational science and engineering. The existing approaches either require explicitly pre-defined basis functions, which makes them inflexible and limits their performance in practical scenarios, or train neural networks as interpolators, which still have limited interpolation accuracy as they treat observed and target points separately and cannot effectively exploit the correlations among data points. Here, we present a learning-based approach to numerical interpolation for scattered data using encoder representation of Transformers (called NIERT). Unlike the recent learning-based approaches, NIERT treats observed and target points in a unified fashion through embedding them into the same representation space, thus gaining the advantage of effectively exploiting the correlations among them. The specially-designed partial self-attention mechanism used by NIERT makes it escape from the unexpected interference of target points on observed points. We further show that the partial self-attention is essentially a learnable interpolation module combining multiple neural basis functions, which provides interpretability of NIERT. Through pre-training on large-scale synthetic datasets, NIERT achieves considerable improvement in interpolation accuracy for practical tasks. On both synthetic and real-world datasets, NIERT outperforms the existing approaches, e.g., on the TFRD-ADlet dataset for temperature field reconstruction, NIERT achieves an MAE of $1.897\times 10^{-3}$, substantially better than the state-of-the-art approach (MAE: $27.074\times 10^{-3}$). The source code of NIERT is available at https://anonymous.4open.science/r/NIERT-2BCF.
Seq-SetNet: Exploring Sequence Sets for Inferring Structures
Jun 06, 2019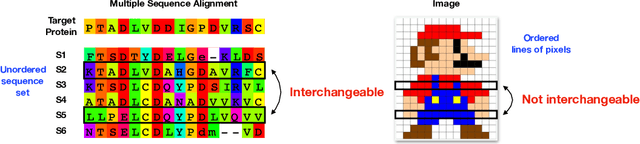
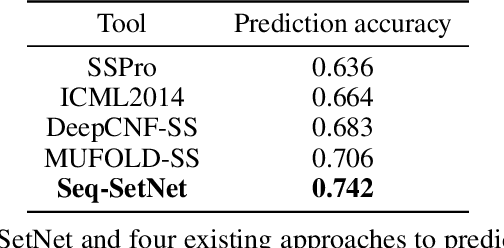
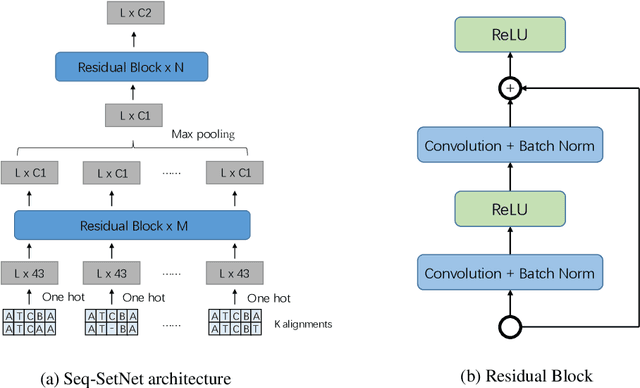
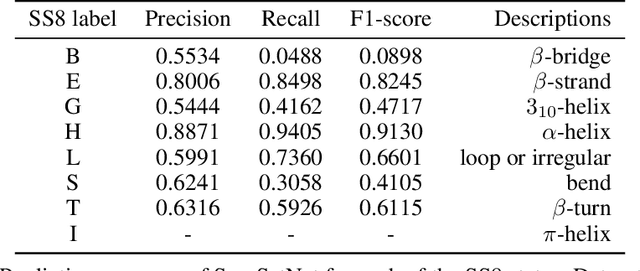
Sequence set is a widely-used type of data source in a large variety of fields. A typical example is protein structure prediction, which takes an multiple sequence alignment (MSA) as input and aims to infer structural information from it. Almost all of the existing approaches exploit MSAs in an indirect fashion, i.e., they transform MSAs into position-specific scoring matrices (PSSM) that represent the distribution of amino acid types at each column. PSSM could capture column-wise characteristics of MSA, however, the column-wise characteristics embedded in each individual component sequence were nearly totally neglected. The drawback of PSSM is rooted in the fact that an MSA is essentially an unordered sequence set rather than a matrix. Specifically, the interchange of any two sequences will not affect the whole MSA. In contrast, the pixels in an image essentially form a matrix since any two rows of pixels cannot be interchanged. Therefore, the traditional deep neural networks designed for image processing cannot be directly applied on sequence sets. Here, we proposed a novel deep neural network framework (called Seq-SetNet) for sequence set processing. By employing a {\it symmetric function} module to integrate features calculated from preceding layers, Seq-SetNet are immune to the order of sequences in the input MSA. This advantage enables us to directly and fully exploit MSAs by considering each component protein individually. We evaluated Seq-SetNet by using it to extract structural information from MSA for protein secondary structure prediction. Experimental results on popular benchmark sets suggests that Seq-SetNet outperforms the state-of-the-art approaches by 3.6% in precision. These results clearly suggest the advantages of Seq-SetNet in sequence set processing and it can be readily used in a wide range of fields, say natural language processing.
Predicting protein inter-residue contacts using composite likelihood maximization and deep learning
Aug 31, 2018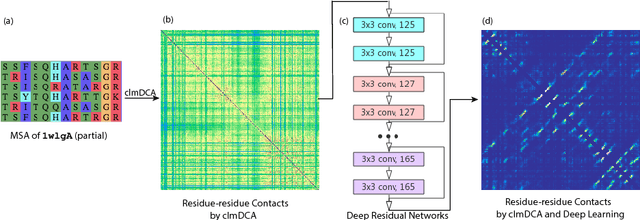
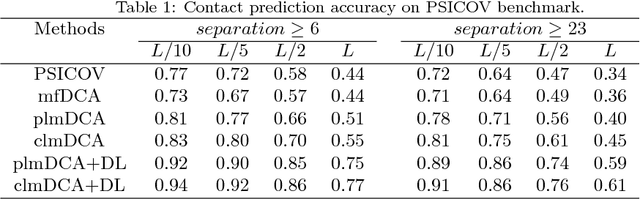
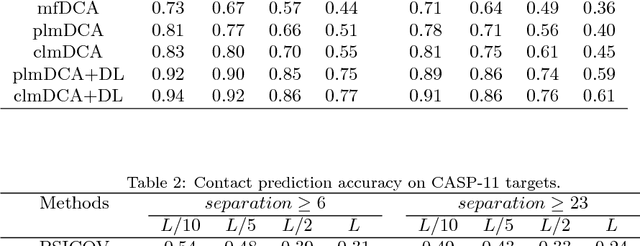
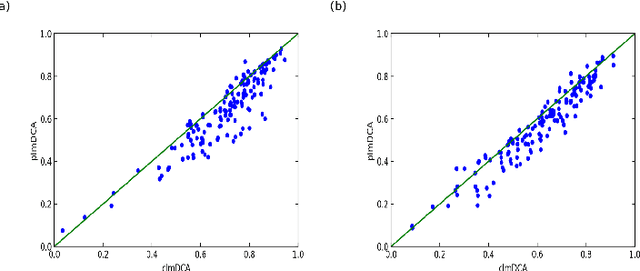
Accurate prediction of inter-residue contacts of a protein is important to calcu- lating its tertiary structure. Analysis of co-evolutionary events among residues has been proved effective to inferring inter-residue contacts. The Markov ran- dom field (MRF) technique, although being widely used for contact prediction, suffers from the following dilemma: the actual likelihood function of MRF is accurate but time-consuming to calculate, in contrast, approximations to the actual likelihood, say pseudo-likelihood, are efficient to calculate but inaccu- rate. Thus, how to achieve both accuracy and efficiency simultaneously remains a challenge. In this study, we present such an approach (called clmDCA) for contact prediction. Unlike plmDCA using pseudo-likelihood, i.e., the product of conditional probability of individual residues, our approach uses composite- likelihood, i.e., the product of conditional probability of all residue pairs. Com- posite likelihood has been theoretically proved as a better approximation to the actual likelihood function than pseudo-likelihood. Meanwhile, composite likelihood is still efficient to maximize, thus ensuring the efficiency of clmDCA. We present comprehensive experiments on popular benchmark datasets, includ- ing PSICOV dataset and CASP-11 dataset, to show that: i) clmDCA alone outperforms the existing MRF-based approaches in prediction accuracy. ii) When equipped with deep learning technique for refinement, the prediction ac- curacy of clmDCA was further significantly improved, suggesting the suitability of clmDCA for subsequent refinement procedure. We further present successful application of the predicted contacts to accurately build tertiary structures for proteins in the PSICOV dataset. Accessibility: The software clmDCA and a server are publicly accessible through http://protein.ict.ac.cn/clmDCA/.