 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDesigning faster mixed integer linear programming algorithm via learning the optimal path
Jan 22, 2026Designing faster algorithms for solving Mixed-Integer Linear Programming (MILP) problems is highly desired across numerous practical domains, as a vast array of complex real-world challenges can be effectively modeled as MILP formulations. Solving these problems typically employs the branch-and-bound algorithm, the core of which can be conceived as searching for a path of nodes (or sub-problems) that contains the optimal solution to the original MILP problem. Traditional approaches to finding this path rely heavily on hand-crafted, intuition-based heuristic strategies, which often suffer from unstable and unpredictable performance across different MILP problem instances. To address this limitation, we introduce DeepBound, a deep learning-based node selection algorithm that automates the learning of such human intuition from data. The core of DeepBound lies in learning to prioritize nodes containing the optimal solution, thereby improving solving efficiency. DeepBound introduces a multi-level feature fusion network to capture the node representations. To tackle the inherent node imbalance in branch-and-bound trees, DeepBound employs a pairwise training paradigm that enhances the model's ability to discriminate between nodes. Extensive experiments on three NP-hard MILP benchmarks demonstrate that DeepBound achieves superior solving efficiency over conventional heuristic rules and existing learning-based approaches, obtaining optimal feasible solutions with significantly reduced computation time. Moreover, DeepBound demonstrates strong generalization capability on large and complex instances. The analysis of its learned features reveals that the method can automatically discover more flexible and robust feature selection, which may effectively improve and potentially replace human-designed heuristic rules.
Predicting mutational effects on protein-protein binding via a side-chain diffusion probabilistic model
Oct 30, 2023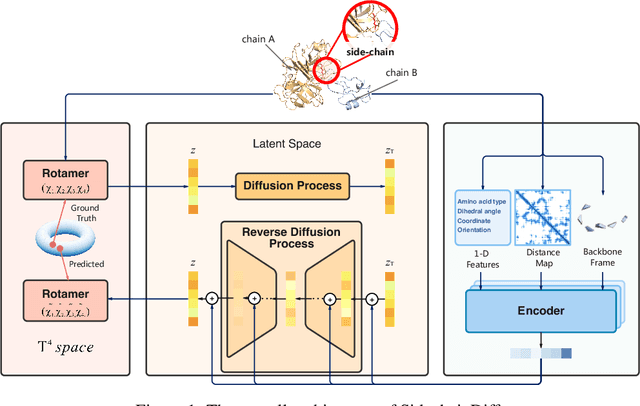
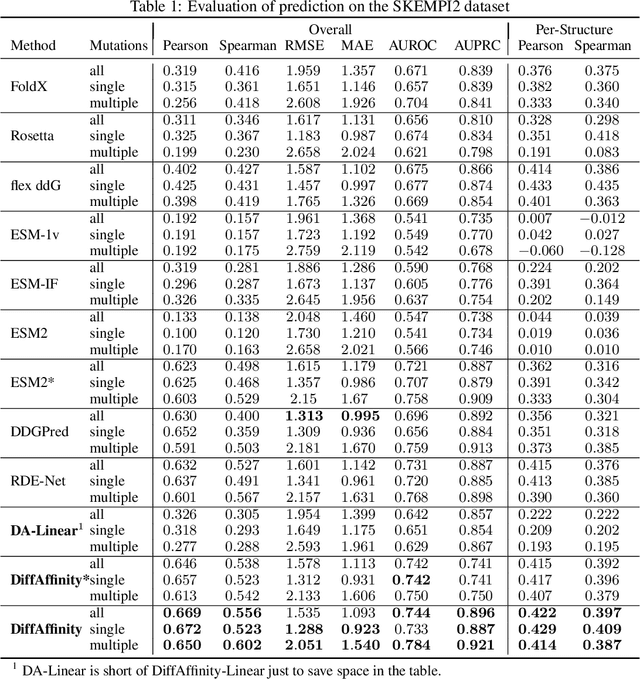
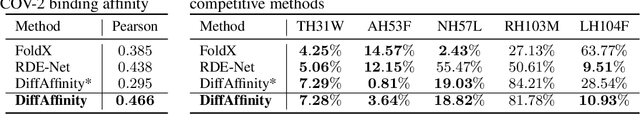
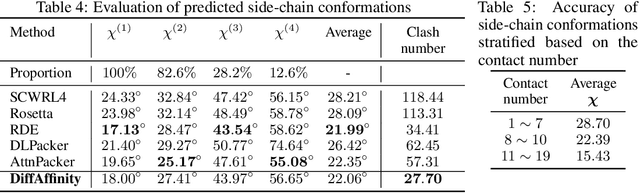
Many crucial biological processes rely on networks of protein-protein interactions. Predicting the effect of amino acid mutations on protein-protein binding is vital in protein engineering and therapeutic discovery. However, the scarcity of annotated experimental data on binding energy poses a significant challenge for developing computational approaches, particularly deep learning-based methods. In this work, we propose SidechainDiff, a representation learning-based approach that leverages unlabelled experimental protein structures. SidechainDiff utilizes a Riemannian diffusion model to learn the generative process of side-chain conformations and can also give the structural context representations of mutations on the protein-protein interface. Leveraging the learned representations, we achieve state-of-the-art performance in predicting the mutational effects on protein-protein binding. Furthermore, SidechainDiff is the first diffusion-based generative model for side-chains, distinguishing it from prior efforts that have predominantly focused on generating protein backbone structures.