 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMulti-scale Graph Autoregressive Modeling: Molecular Property Prediction via Next Token Prediction
Jan 05, 2026We present Connection-Aware Motif Sequencing (CamS), a graph-to-sequence representation that enables decoder-only Transformers to learn molecular graphs via standard next-token prediction (NTP). For molecular property prediction, SMILES-based NTP scales well but lacks explicit topology, whereas graph-native masked modeling captures connectivity but risks disrupting the pivotal chemical details (e.g., activity cliffs). CamS bridges this gap by serializing molecular graphs into structure-rich causal sequences. CamS first mines data-driven connection-aware motifs. It then serializes motifs via scaffold-rooted breadth-first search (BFS) to establish a stable core-to-periphery order. Crucially, CamS enables hierarchical modeling by concatenating sequences from fine to coarse motif scales, allowing the model to condition global scaffolds on dense, uncorrupted local structural evidence. We instantiate CamS-LLaMA by pre-training a vanilla LLaMA backbone on CamS sequences. It achieves state-of-the-art performance on MoleculeNet and the activity-cliff benchmark MoleculeACE, outperforming both SMILES-based language models and strong graph baselines. Interpretability analysis confirms that our multi-scale causal serialization effectively drives attention toward cliff-determining differences.
MolChord: Structure-Sequence Alignment for Protein-Guided Drug Design
Oct 31, 2025Structure-based drug design (SBDD), which maps target proteins to candidate molecular ligands, is a fundamental task in drug discovery. Effectively aligning protein structural representations with molecular representations, and ensuring alignment between generated drugs and their pharmacological properties, remains a critical challenge. To address these challenges, we propose MolChord, which integrates two key techniques: (1) to align protein and molecule structures with their textual descriptions and sequential representations (e.g., FASTA for proteins and SMILES for molecules), we leverage NatureLM, an autoregressive model unifying text, small molecules, and proteins, as the molecule generator, alongside a diffusion-based structure encoder; and (2) to guide molecules toward desired properties, we curate a property-aware dataset by integrating preference data and refine the alignment process using Direct Preference Optimization (DPO). Experimental results on CrossDocked2020 demonstrate that our approach achieves state-of-the-art performance on key evaluation metrics, highlighting its potential as a practical tool for SBDD.
Trust Region Preference Approximation: A simple and stable reinforcement learning algorithm for LLM reasoning
Apr 06, 2025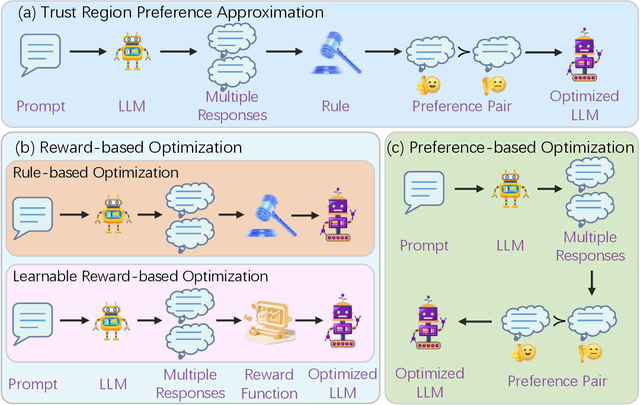
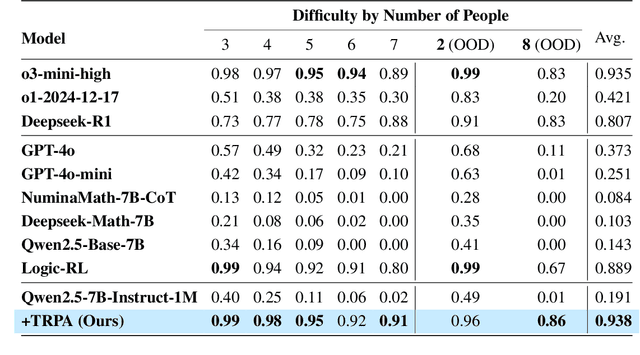
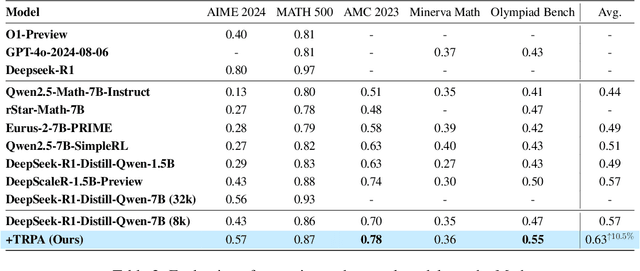
Recently, Large Language Models (LLMs) have rapidly evolved, approaching Artificial General Intelligence (AGI) while benefiting from large-scale reinforcement learning to enhance Human Alignment (HA) and Reasoning. Recent reward-based optimization algorithms, such as Proximal Policy Optimization (PPO) and Group Relative Policy Optimization (GRPO) have achieved significant performance on reasoning tasks, whereas preference-based optimization algorithms such as Direct Preference Optimization (DPO) significantly improve the performance of LLMs on human alignment. However, despite the strong performance of reward-based optimization methods in alignment tasks , they remain vulnerable to reward hacking. Furthermore, preference-based algorithms (such as Online DPO) haven't yet matched the performance of reward-based optimization algorithms (like PPO) on reasoning tasks, making their exploration in this specific area still a worthwhile pursuit. Motivated by these challenges, we propose the Trust Region Preference Approximation (TRPA) algorithm, which integrates rule-based optimization with preference-based optimization for reasoning tasks. As a preference-based algorithm, TRPA naturally eliminates the reward hacking issue. TRPA constructs preference levels using predefined rules, forms corresponding preference pairs, and leverages a novel optimization algorithm for RL training with a theoretical monotonic improvement guarantee. Experimental results demonstrate that TRPA not only achieves competitive performance on reasoning tasks but also exhibits robust stability. The code of this paper are released and updating on https://github.com/XueruiSu/Trust-Region-Preference-Approximation.git.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
\underline{E2}Former: A Linear-time \underline{E}fficient and \underline{E}quivariant Trans\underline{former} for Scalable Molecular Modeling
Jan 31, 2025



Equivariant Graph Neural Networks (EGNNs) have demonstrated significant success in modeling microscale systems, including those in chemistry, biology and materials science. However, EGNNs face substantial computational challenges due to the high cost of constructing edge features via spherical tensor products, making them impractical for large-scale systems. To address this limitation, we introduce E2Former, an equivariant and efficient transformer architecture that incorporates the Wigner $6j$ convolution (Wigner $6j$ Conv). By shifting the computational burden from edges to nodes, the Wigner $6j$ Conv reduces the complexity from $O(|\mathcal{E}|)$ to $ O(| \mathcal{V}|)$ while preserving both the model's expressive power and rotational equivariance. We show that this approach achieves a 7x-30x speedup compared to conventional $\mathrm{SO}(3)$ convolutions. Furthermore, our empirical results demonstrate that the derived E2Former mitigates the computational challenges of existing approaches without compromising the ability to capture detailed geometric information. This development could suggest a promising direction for scalable and efficient molecular modeling.
SFM-Protein: Integrative Co-evolutionary Pre-training for Advanced Protein Sequence Representation
Oct 31, 2024



Proteins, essential to biological systems, perform functions intricately linked to their three-dimensional structures. Understanding the relationship between protein structures and their amino acid sequences remains a core challenge in protein modeling. While traditional protein foundation models benefit from pre-training on vast unlabeled datasets, they often struggle to capture critical co-evolutionary information, which evolutionary-based methods excel at. In this study, we introduce a novel pre-training strategy for protein foundation models that emphasizes the interactions among amino acid residues to enhance the extraction of both short-range and long-range co-evolutionary features from sequence data. Trained on a large-scale protein sequence dataset, our model demonstrates superior generalization ability, outperforming established baselines of similar size, including the ESM model, across diverse downstream tasks. Experimental results confirm the model's effectiveness in integrating co-evolutionary information, marking a significant step forward in protein sequence-based modeling.
Physical Consistency Bridges Heterogeneous Data in Molecular Multi-Task Learning
Oct 14, 2024



In recent years, machine learning has demonstrated impressive capability in handling molecular science tasks. To support various molecular properties at scale, machine learning models are trained in the multi-task learning paradigm. Nevertheless, data of different molecular properties are often not aligned: some quantities, e.g. equilibrium structure, demand more cost to compute than others, e.g. energy, so their data are often generated by cheaper computational methods at the cost of lower accuracy, which cannot be directly overcome through multi-task learning. Moreover, it is not straightforward to leverage abundant data of other tasks to benefit a particular task. To handle such data heterogeneity challenges, we exploit the specialty of molecular tasks that there are physical laws connecting them, and design consistency training approaches that allow different tasks to exchange information directly so as to improve one another. Particularly, we demonstrate that the more accurate energy data can improve the accuracy of structure prediction. We also find that consistency training can directly leverage force and off-equilibrium structure data to improve structure prediction, demonstrating a broad capability for integrating heterogeneous data.
Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
Jun 08, 2023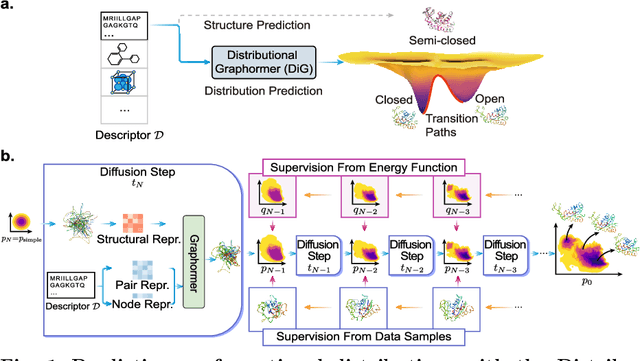
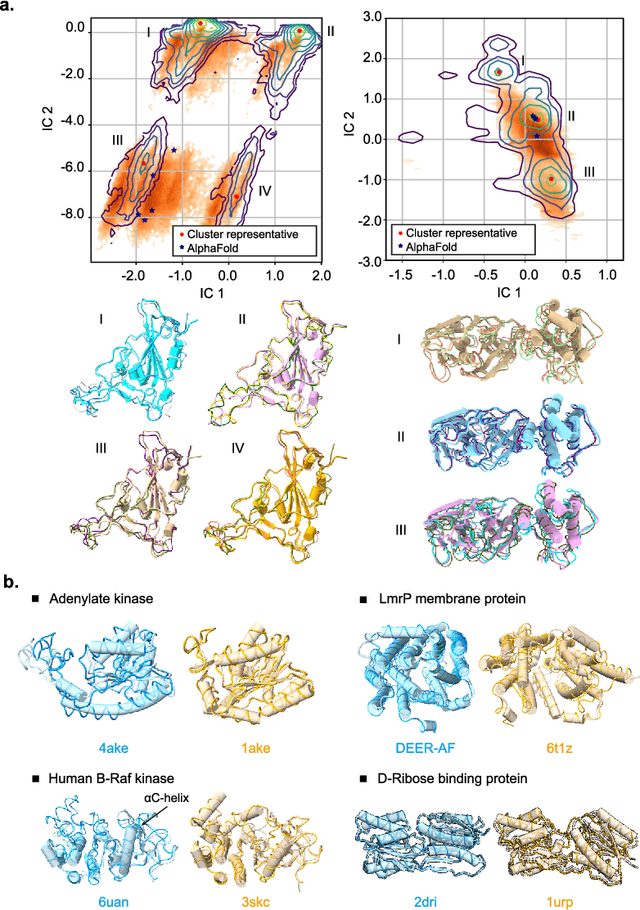
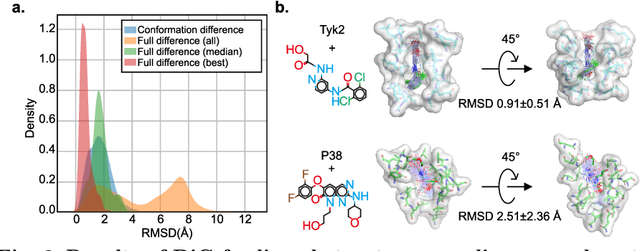
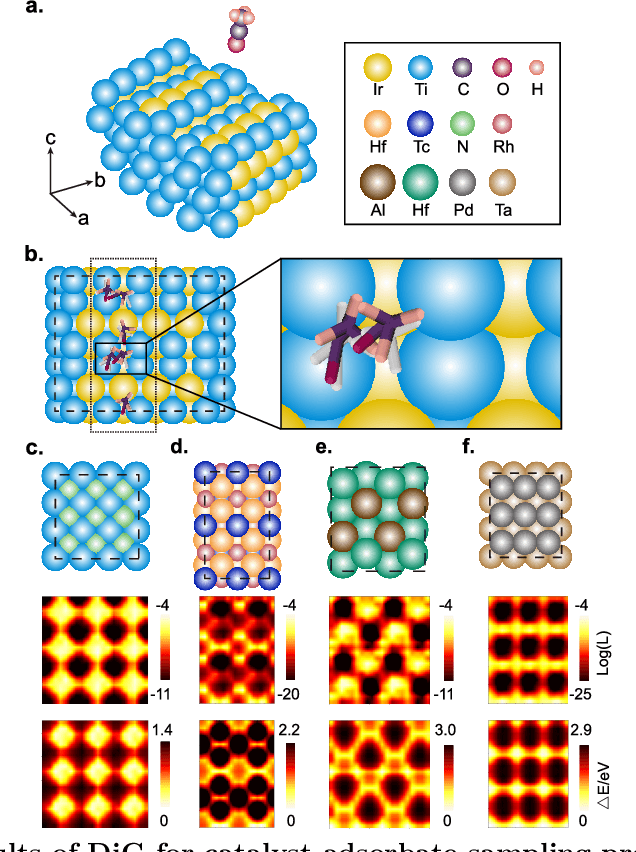
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.