 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeQwen-Image-Layered: Towards Inherent Editability via Layer Decomposition
Dec 17, 2025Recent visual generative models often struggle with consistency during image editing due to the entangled nature of raster images, where all visual content is fused into a single canvas. In contrast, professional design tools employ layered representations, allowing isolated edits while preserving consistency. Motivated by this, we propose \textbf{Qwen-Image-Layered}, an end-to-end diffusion model that decomposes a single RGB image into multiple semantically disentangled RGBA layers, enabling \textbf{inherent editability}, where each RGBA layer can be independently manipulated without affecting other content. To support variable-length decomposition, we introduce three key components: (1) an RGBA-VAE to unify the latent representations of RGB and RGBA images; (2) a VLD-MMDiT (Variable Layers Decomposition MMDiT) architecture capable of decomposing a variable number of image layers; and (3) a Multi-stage Training strategy to adapt a pretrained image generation model into a multilayer image decomposer. Furthermore, to address the scarcity of high-quality multilayer training images, we build a pipeline to extract and annotate multilayer images from Photoshop documents (PSD). Experiments demonstrate that our method significantly surpasses existing approaches in decomposition quality and establishes a new paradigm for consistent image editing. Our code and models are released on \href{https://github.com/QwenLM/Qwen-Image-Layered}{https://github.com/QwenLM/Qwen-Image-Layered}
UniGenX: Unified Generation of Sequence and Structure with Autoregressive Diffusion
Mar 09, 2025Unified generation of sequence and structure for scientific data (e.g., materials, molecules, proteins) is a critical task. Existing approaches primarily rely on either autoregressive sequence models or diffusion models, each offering distinct advantages and facing notable limitations. Autoregressive models, such as GPT, Llama, and Phi-4, have demonstrated remarkable success in natural language generation and have been extended to multimodal tasks (e.g., image, video, and audio) using advanced encoders like VQ-VAE to represent complex modalities as discrete sequences. However, their direct application to scientific domains is challenging due to the high precision requirements and the diverse nature of scientific data. On the other hand, diffusion models excel at generating high-dimensional scientific data, such as protein, molecule, and material structures, with remarkable accuracy. Yet, their inability to effectively model sequences limits their potential as general-purpose multimodal foundation models. To address these challenges, we propose UniGenX, a unified framework that combines autoregressive next-token prediction with conditional diffusion models. This integration leverages the strengths of autoregressive models to ease the training of conditional diffusion models, while diffusion-based generative heads enhance the precision of autoregressive predictions. We validate the effectiveness of UniGenX on material and small molecule generation tasks, achieving a significant leap in state-of-the-art performance for material crystal structure prediction and establishing new state-of-the-art results for small molecule structure prediction, de novo design, and conditional generation. Notably, UniGenX demonstrates significant improvements, especially in handling long sequences for complex structures, showcasing its efficacy as a versatile tool for scientific data generation.
Fast and Accurate Blind Flexible Docking
Feb 20, 2025Molecular docking that predicts the bound structures of small molecules (ligands) to their protein targets, plays a vital role in drug discovery. However, existing docking methods often face limitations: they either overlook crucial structural changes by assuming protein rigidity or suffer from low computational efficiency due to their reliance on generative models for structure sampling. To address these challenges, we propose FABFlex, a fast and accurate regression-based multi-task learning model designed for realistic blind flexible docking scenarios, where proteins exhibit flexibility and binding pocket sites are unknown (blind). Specifically, FABFlex's architecture comprises three specialized modules working in concert: (1) A pocket prediction module that identifies potential binding sites, addressing the challenges inherent in blind docking scenarios. (2) A ligand docking module that predicts the bound (holo) structures of ligands from their unbound (apo) states. (3) A pocket docking module that forecasts the holo structures of protein pockets from their apo conformations. Notably, FABFlex incorporates an iterative update mechanism that serves as a conduit between the ligand and pocket docking modules, enabling continuous structural refinements. This approach effectively integrates the three subtasks of blind flexible docking-pocket identification, ligand conformation prediction, and protein flexibility modeling-into a unified, coherent framework. Extensive experiments on public benchmark datasets demonstrate that FABFlex not only achieves superior effectiveness in predicting accurate binding modes but also exhibits a significant speed advantage (208 $\times$) compared to existing state-of-the-art methods. Our code is released at https://github.com/tmlr-group/FABFlex.
NatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Tokenizing 3D Molecule Structure with Quantized Spherical Coordinates
Dec 02, 2024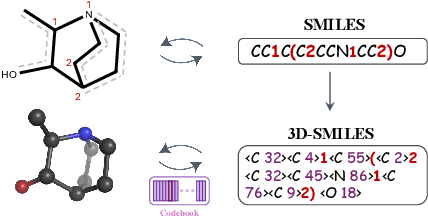
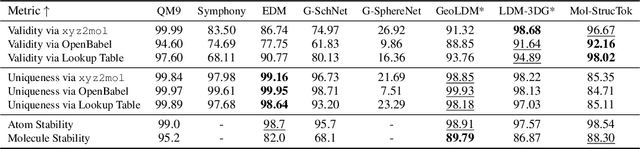
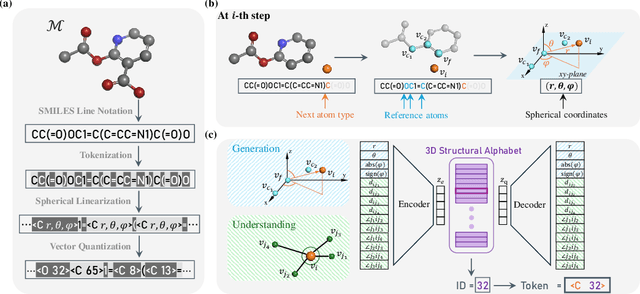
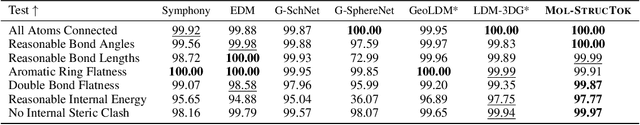
The application of language models (LMs) to molecular structure generation using line notations such as SMILES and SELFIES has been well-established in the field of cheminformatics. However, extending these models to generate 3D molecular structures presents significant challenges. Two primary obstacles emerge: (1) the difficulty in designing a 3D line notation that ensures SE(3)-invariant atomic coordinates, and (2) the non-trivial task of tokenizing continuous coordinates for use in LMs, which inherently require discrete inputs. To address these challenges, we propose Mol-StrucTok, a novel method for tokenizing 3D molecular structures. Our approach comprises two key innovations: (1) We design a line notation for 3D molecules by extracting local atomic coordinates in a spherical coordinate system. This notation builds upon existing 2D line notations and remains agnostic to their specific forms, ensuring compatibility with various molecular representation schemes. (2) We employ a Vector Quantized Variational Autoencoder (VQ-VAE) to tokenize these coordinates, treating them as generation descriptors. To further enhance the representation, we incorporate neighborhood bond lengths and bond angles as understanding descriptors. Leveraging this tokenization framework, we train a GPT-2 style model for 3D molecular generation tasks. Results demonstrate strong performance with significantly faster generation speeds and competitive chemical stability compared to previous methods. Further, by integrating our learned discrete representations into Graphormer model for property prediction on QM9 dataset, Mol-StrucTok reveals consistent improvements across various molecular properties, underscoring the versatility and robustness of our approach.
SFM-Protein: Integrative Co-evolutionary Pre-training for Advanced Protein Sequence Representation
Oct 31, 2024



Proteins, essential to biological systems, perform functions intricately linked to their three-dimensional structures. Understanding the relationship between protein structures and their amino acid sequences remains a core challenge in protein modeling. While traditional protein foundation models benefit from pre-training on vast unlabeled datasets, they often struggle to capture critical co-evolutionary information, which evolutionary-based methods excel at. In this study, we introduce a novel pre-training strategy for protein foundation models that emphasizes the interactions among amino acid residues to enhance the extraction of both short-range and long-range co-evolutionary features from sequence data. Trained on a large-scale protein sequence dataset, our model demonstrates superior generalization ability, outperforming established baselines of similar size, including the ESM model, across diverse downstream tasks. Experimental results confirm the model's effectiveness in integrating co-evolutionary information, marking a significant step forward in protein sequence-based modeling.
3D-MolT5: Towards Unified 3D Molecule-Text Modeling with 3D Molecular Tokenization
Jun 09, 2024



The integration of molecule and language has garnered increasing attention in molecular science. Recent advancements in Language Models (LMs) have demonstrated potential for the comprehensive modeling of molecule and language. However, existing works exhibit notable limitations. Most existing works overlook the modeling of 3D information, which is crucial for understanding molecular structures and also functions. While some attempts have been made to leverage external structure encoding modules to inject the 3D molecular information into LMs, there exist obvious difficulties that hinder the integration of molecular structure and language text, such as modality alignment and separate tuning. To bridge this gap, we propose 3D-MolT5, a unified framework designed to model both 1D molecular sequence and 3D molecular structure. The key innovation lies in our methodology for mapping fine-grained 3D substructure representations (based on 3D molecular fingerprints) to a specialized 3D token vocabulary for 3D-MolT5. This 3D structure token vocabulary enables the seamless combination of 1D sequence and 3D structure representations in a tokenized format, allowing 3D-MolT5 to encode molecular sequence (SELFIES), molecular structure, and text sequences within a unified architecture. Alongside, we further introduce 1D and 3D joint pre-training to enhance the model's comprehension of these diverse modalities in a joint representation space and better generalize to various tasks for our foundation model. Through instruction tuning on multiple downstream datasets, our proposed 3D-MolT5 shows superior performance than existing methods in molecular property prediction, molecule captioning, and text-based molecule generation tasks. Our code will be available on GitHub soon.
FABind+: Enhancing Molecular Docking through Improved Pocket Prediction and Pose Generation
Apr 07, 2024



Molecular docking is a pivotal process in drug discovery. While traditional techniques rely on extensive sampling and simulation governed by physical principles, these methods are often slow and costly. The advent of deep learning-based approaches has shown significant promise, offering increases in both accuracy and efficiency. Building upon the foundational work of FABind, a model designed with a focus on speed and accuracy, we present FABind+, an enhanced iteration that largely boosts the performance of its predecessor. We identify pocket prediction as a critical bottleneck in molecular docking and propose a novel methodology that significantly refines pocket prediction, thereby streamlining the docking process. Furthermore, we introduce modifications to the docking module to enhance its pose generation capabilities. In an effort to bridge the gap with conventional sampling/generative methods, we incorporate a simple yet effective sampling technique coupled with a confidence model, requiring only minor adjustments to the regression framework of FABind. Experimental results and analysis reveal that FABind+ remarkably outperforms the original FABind, achieves competitive state-of-the-art performance, and delivers insightful modeling strategies. This demonstrates FABind+ represents a substantial step forward in molecular docking and drug discovery. Our code is in https://github.com/QizhiPei/FABind.
Leveraging Biomolecule and Natural Language through Multi-Modal Learning: A Survey
Mar 05, 2024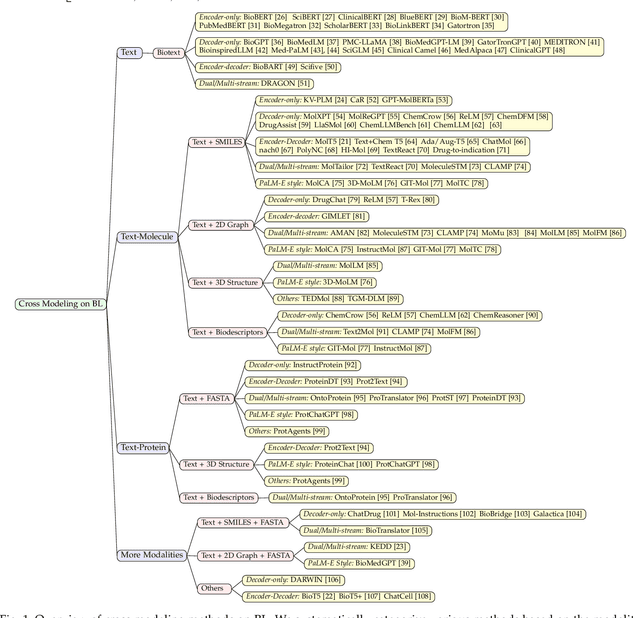



The integration of biomolecular modeling with natural language (BL) has emerged as a promising interdisciplinary area at the intersection of artificial intelligence, chemistry and biology. This approach leverages the rich, multifaceted descriptions of biomolecules contained within textual data sources to enhance our fundamental understanding and enable downstream computational tasks such as biomolecule property prediction. The fusion of the nuanced narratives expressed through natural language with the structural and functional specifics of biomolecules described via various molecular modeling techniques opens new avenues for comprehensively representing and analyzing biomolecules. By incorporating the contextual language data that surrounds biomolecules into their modeling, BL aims to capture a holistic view encompassing both the symbolic qualities conveyed through language as well as quantitative structural characteristics. In this review, we provide an extensive analysis of recent advancements achieved through cross modeling of biomolecules and natural language. (1) We begin by outlining the technical representations of biomolecules employed, including sequences, 2D graphs, and 3D structures. (2) We then examine in depth the rationale and key objectives underlying effective multi-modal integration of language and molecular data sources. (3) We subsequently survey the practical applications enabled to date in this developing research area. (4) We also compile and summarize the available resources and datasets to facilitate future work. (5) Looking ahead, we identify several promising research directions worthy of further exploration and investment to continue advancing the field. The related resources and contents are updating in \url{https://github.com/QizhiPei/Awesome-Biomolecule-Language-Cross-Modeling}.
BioT5+: Towards Generalized Biological Understanding with IUPAC Integration and Multi-task Tuning
Feb 27, 2024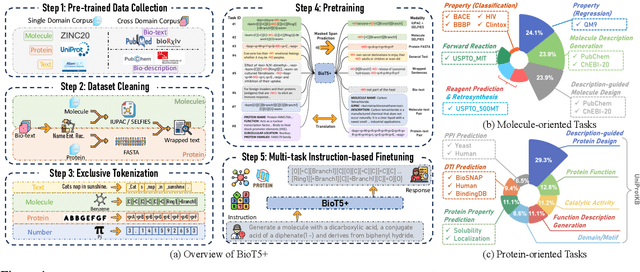
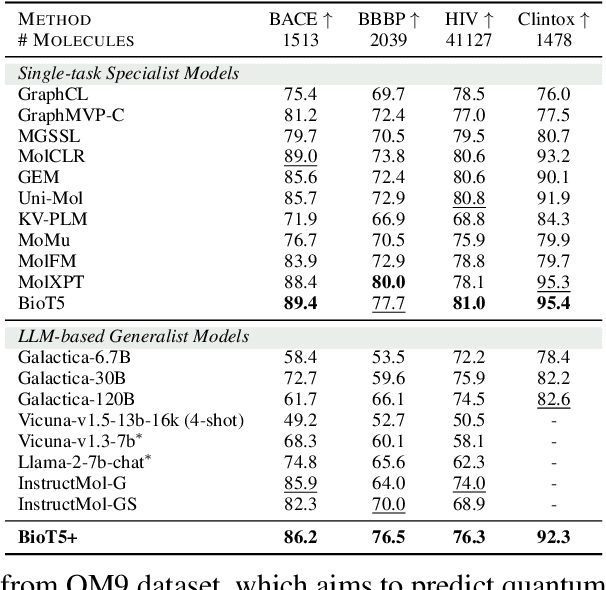
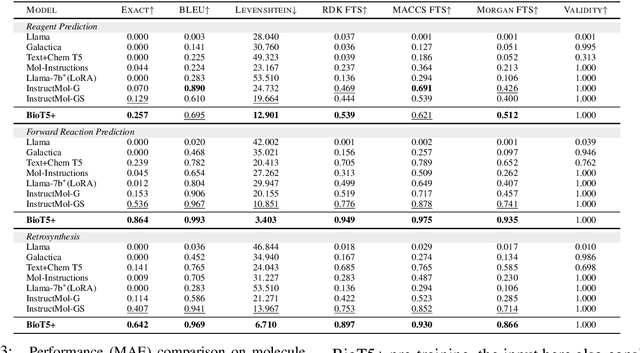
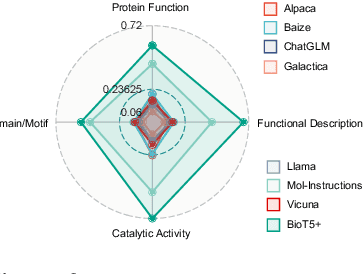
Recent research trends in computational biology have increasingly focused on integrating text and bio-entity modeling, especially in the context of molecules and proteins. However, previous efforts like BioT5 faced challenges in generalizing across diverse tasks and lacked a nuanced understanding of molecular structures, particularly in their textual representations (e.g., IUPAC). This paper introduces BioT5+, an extension of the BioT5 framework, tailored to enhance biological research and drug discovery. BioT5+ incorporates several novel features: integration of IUPAC names for molecular understanding, inclusion of extensive bio-text and molecule data from sources like bioRxiv and PubChem, the multi-task instruction tuning for generality across tasks, and a novel numerical tokenization technique for improved processing of numerical data. These enhancements allow BioT5+ to bridge the gap between molecular representations and their textual descriptions, providing a more holistic understanding of biological entities, and largely improving the grounded reasoning of bio-text and bio-sequences. The model is pre-trained and fine-tuned with a large number of experiments, including \emph{3 types of problems (classification, regression, generation), 15 kinds of tasks, and 21 total benchmark datasets}, demonstrating the remarkable performance and state-of-the-art results in most cases. BioT5+ stands out for its ability to capture intricate relationships in biological data, thereby contributing significantly to bioinformatics and computational biology. Our code is available at \url{https://github.com/QizhiPei/BioT5}.