 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNatureLM: Deciphering the Language of Nature for Scientific Discovery
Feb 11, 2025



Foundation models have revolutionized natural language processing and artificial intelligence, significantly enhancing how machines comprehend and generate human languages. Inspired by the success of these foundation models, researchers have developed foundation models for individual scientific domains, including small molecules, materials, proteins, DNA, and RNA. However, these models are typically trained in isolation, lacking the ability to integrate across different scientific domains. Recognizing that entities within these domains can all be represented as sequences, which together form the "language of nature", we introduce Nature Language Model (briefly, NatureLM), a sequence-based science foundation model designed for scientific discovery. Pre-trained with data from multiple scientific domains, NatureLM offers a unified, versatile model that enables various applications including: (i) generating and optimizing small molecules, proteins, RNA, and materials using text instructions; (ii) cross-domain generation/design, such as protein-to-molecule and protein-to-RNA generation; and (iii) achieving state-of-the-art performance in tasks like SMILES-to-IUPAC translation and retrosynthesis on USPTO-50k. NatureLM offers a promising generalist approach for various scientific tasks, including drug discovery (hit generation/optimization, ADMET optimization, synthesis), novel material design, and the development of therapeutic proteins or nucleotides. We have developed NatureLM models in different sizes (1 billion, 8 billion, and 46.7 billion parameters) and observed a clear improvement in performance as the model size increases.
Bridging Geometric States via Geometric Diffusion Bridge
Oct 31, 2024

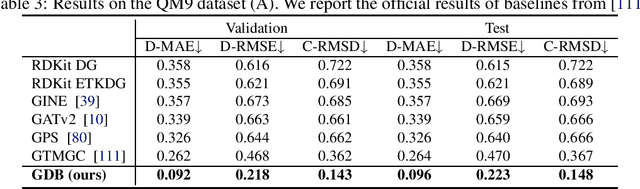
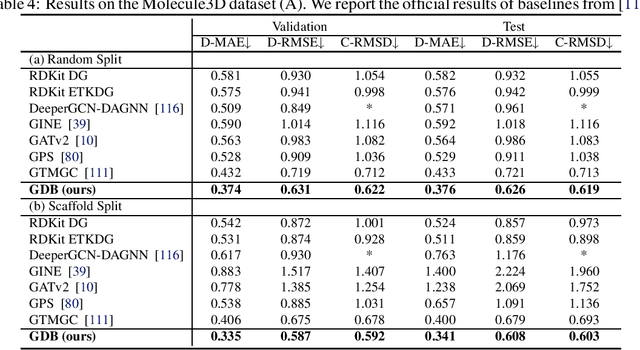
The accurate prediction of geometric state evolution in complex systems is critical for advancing scientific domains such as quantum chemistry and material modeling. Traditional experimental and computational methods face challenges in terms of environmental constraints and computational demands, while current deep learning approaches still fall short in terms of precision and generality. In this work, we introduce the Geometric Diffusion Bridge (GDB), a novel generative modeling framework that accurately bridges initial and target geometric states. GDB leverages a probabilistic approach to evolve geometric state distributions, employing an equivariant diffusion bridge derived by a modified version of Doob's $h$-transform for connecting geometric states. This tailored diffusion process is anchored by initial and target geometric states as fixed endpoints and governed by equivariant transition kernels. Moreover, trajectory data can be seamlessly leveraged in our GDB framework by using a chain of equivariant diffusion bridges, providing a more detailed and accurate characterization of evolution dynamics. Theoretically, we conduct a thorough examination to confirm our framework's ability to preserve joint distributions of geometric states and capability to completely model the underlying dynamics inducing trajectory distributions with negligible error. Experimental evaluations across various real-world scenarios show that GDB surpasses existing state-of-the-art approaches, opening up a new pathway for accurately bridging geometric states and tackling crucial scientific challenges with improved accuracy and applicability.
SFM-Protein: Integrative Co-evolutionary Pre-training for Advanced Protein Sequence Representation
Oct 31, 2024



Proteins, essential to biological systems, perform functions intricately linked to their three-dimensional structures. Understanding the relationship between protein structures and their amino acid sequences remains a core challenge in protein modeling. While traditional protein foundation models benefit from pre-training on vast unlabeled datasets, they often struggle to capture critical co-evolutionary information, which evolutionary-based methods excel at. In this study, we introduce a novel pre-training strategy for protein foundation models that emphasizes the interactions among amino acid residues to enhance the extraction of both short-range and long-range co-evolutionary features from sequence data. Trained on a large-scale protein sequence dataset, our model demonstrates superior generalization ability, outperforming established baselines of similar size, including the ESM model, across diverse downstream tasks. Experimental results confirm the model's effectiveness in integrating co-evolutionary information, marking a significant step forward in protein sequence-based modeling.
Physical Consistency Bridges Heterogeneous Data in Molecular Multi-Task Learning
Oct 14, 2024



In recent years, machine learning has demonstrated impressive capability in handling molecular science tasks. To support various molecular properties at scale, machine learning models are trained in the multi-task learning paradigm. Nevertheless, data of different molecular properties are often not aligned: some quantities, e.g. equilibrium structure, demand more cost to compute than others, e.g. energy, so their data are often generated by cheaper computational methods at the cost of lower accuracy, which cannot be directly overcome through multi-task learning. Moreover, it is not straightforward to leverage abundant data of other tasks to benefit a particular task. To handle such data heterogeneity challenges, we exploit the specialty of molecular tasks that there are physical laws connecting them, and design consistency training approaches that allow different tasks to exchange information directly so as to improve one another. Particularly, we demonstrate that the more accurate energy data can improve the accuracy of structure prediction. We also find that consistency training can directly leverage force and off-equilibrium structure data to improve structure prediction, demonstrating a broad capability for integrating heterogeneous data.
Pattern based learning and optimisation through pricing for bin packing problem
Aug 27, 2024



As a popular form of knowledge and experience, patterns and their identification have been critical tasks in most data mining applications. However, as far as we are aware, no study has systematically examined the dynamics of pattern values and their reuse under varying conditions. We argue that when problem conditions such as the distributions of random variables change, the patterns that performed well in previous circumstances may become less effective and adoption of these patterns would result in sub-optimal solutions. In response, we make a connection between data mining and the duality theory in operations research and propose a novel scheme to efficiently identify patterns and dynamically quantify their values for each specific condition. Our method quantifies the value of patterns based on their ability to satisfy stochastic constraints and their effects on the objective value, allowing high-quality patterns and their combinations to be detected. We use the online bin packing problem to evaluate the effectiveness of the proposed scheme and illustrate the online packing procedure with the guidance of patterns that address the inherent uncertainty of the problem. Results show that the proposed algorithm significantly outperforms the state-of-the-art methods. We also analysed in detail the distinctive features of the proposed methods that lead to performance improvement and the special cases where our method can be further improved.
GeoMFormer: A General Architecture for Geometric Molecular Representation Learning
Jun 24, 2024Molecular modeling, a central topic in quantum mechanics, aims to accurately calculate the properties and simulate the behaviors of molecular systems. The molecular model is governed by physical laws, which impose geometric constraints such as invariance and equivariance to coordinate rotation and translation. While numerous deep learning approaches have been developed to learn molecular representations under these constraints, most of them are built upon heuristic and costly modules. We argue that there is a strong need for a general and flexible framework for learning both invariant and equivariant features. In this work, we introduce a novel Transformer-based molecular model called GeoMFormer to achieve this goal. Using the standard Transformer modules, two separate streams are developed to maintain and learn invariant and equivariant representations. Carefully designed cross-attention modules bridge the two streams, allowing information fusion and enhancing geometric modeling in each stream. As a general and flexible architecture, we show that many previous architectures can be viewed as special instantiations of GeoMFormer. Extensive experiments are conducted to demonstrate the power of GeoMFormer. All empirical results show that GeoMFormer achieves strong performance on both invariant and equivariant tasks of different types and scales. Code and models will be made publicly available at https://github.com/c-tl/GeoMFormer.
Self-Consistency Training for Hamiltonian Prediction
Mar 14, 2024



Hamiltonian prediction is a versatile formulation to leverage machine learning for solving molecular science problems. Yet, its applicability is limited by insufficient labeled data for training. In this work, we highlight that Hamiltonian prediction possesses a self-consistency principle, based on which we propose an exact training method that does not require labeled data. This merit addresses the data scarcity difficulty, and distinguishes the task from other property prediction formulations with unique benefits: (1) self-consistency training enables the model to be trained on a large amount of unlabeled data, hence substantially enhances generalization; (2) self-consistency training is more efficient than labeling data with DFT for supervised training, since it is an amortization of DFT calculation over a set of molecular structures. We empirically demonstrate the better generalization in data-scarce and out-of-distribution scenarios, and the better efficiency from the amortization. These benefits push forward the applicability of Hamiltonian prediction to an ever larger scale.
FABind: Fast and Accurate Protein-Ligand Binding
Oct 17, 2023Modeling the interaction between proteins and ligands and accurately predicting their binding structures is a critical yet challenging task in drug discovery. Recent advancements in deep learning have shown promise in addressing this challenge, with sampling-based and regression-based methods emerging as two prominent approaches. However, these methods have notable limitations. Sampling-based methods often suffer from low efficiency due to the need for generating multiple candidate structures for selection. On the other hand, regression-based methods offer fast predictions but may experience decreased accuracy. Additionally, the variation in protein sizes often requires external modules for selecting suitable binding pockets, further impacting efficiency. In this work, we propose $\mathbf{FABind}$, an end-to-end model that combines pocket prediction and docking to achieve accurate and fast protein-ligand binding. $\mathbf{FABind}$ incorporates a unique ligand-informed pocket prediction module, which is also leveraged for docking pose estimation. The model further enhances the docking process by incrementally integrating the predicted pocket to optimize protein-ligand binding, reducing discrepancies between training and inference. Through extensive experiments on benchmark datasets, our proposed $\mathbf{FABind}$ demonstrates strong advantages in terms of effectiveness and efficiency compared to existing methods. Our code is available at $\href{https://github.com/QizhiPei/FABind}{Github}$.
Learning Multi-Agent Intention-Aware Communication for Optimal Multi-Order Execution in Finance
Jul 06, 2023
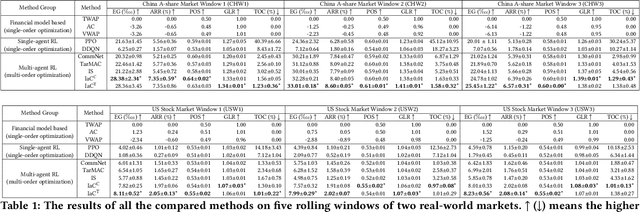


Order execution is a fundamental task in quantitative finance, aiming at finishing acquisition or liquidation for a number of trading orders of the specific assets. Recent advance in model-free reinforcement learning (RL) provides a data-driven solution to the order execution problem. However, the existing works always optimize execution for an individual order, overlooking the practice that multiple orders are specified to execute simultaneously, resulting in suboptimality and bias. In this paper, we first present a multi-agent RL (MARL) method for multi-order execution considering practical constraints. Specifically, we treat every agent as an individual operator to trade one specific order, while keeping communicating with each other and collaborating for maximizing the overall profits. Nevertheless, the existing MARL algorithms often incorporate communication among agents by exchanging only the information of their partial observations, which is inefficient in complicated financial market. To improve collaboration, we then propose a learnable multi-round communication protocol, for the agents communicating the intended actions with each other and refining accordingly. It is optimized through a novel action value attribution method which is provably consistent with the original learning objective yet more efficient. The experiments on the data from two real-world markets have illustrated superior performance with significantly better collaboration effectiveness achieved by our method.
Towards Predicting Equilibrium Distributions for Molecular Systems with Deep Learning
Jun 08, 2023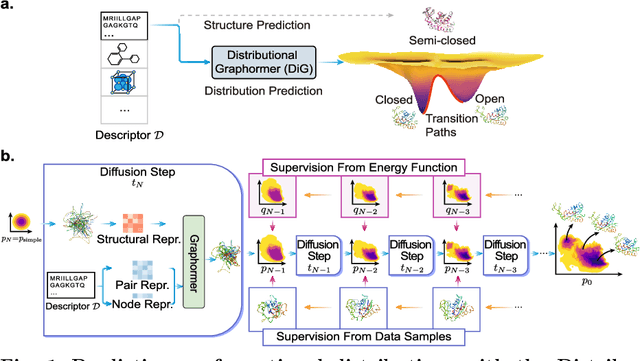
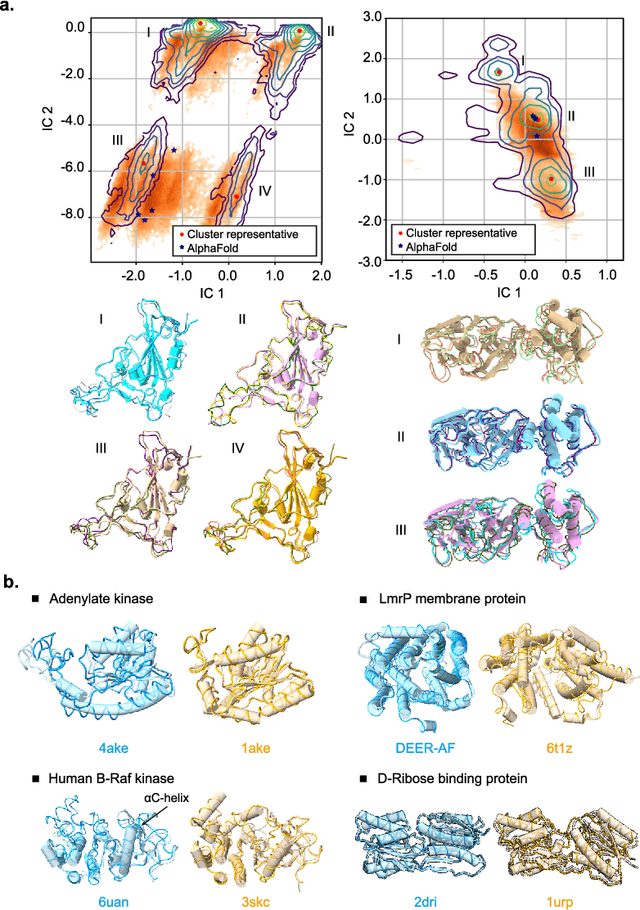
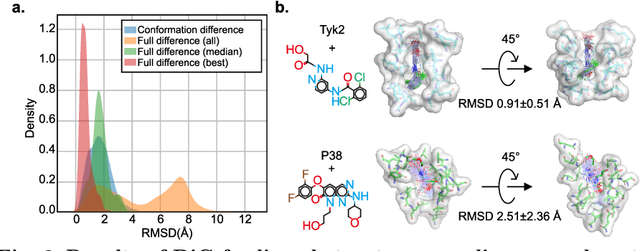
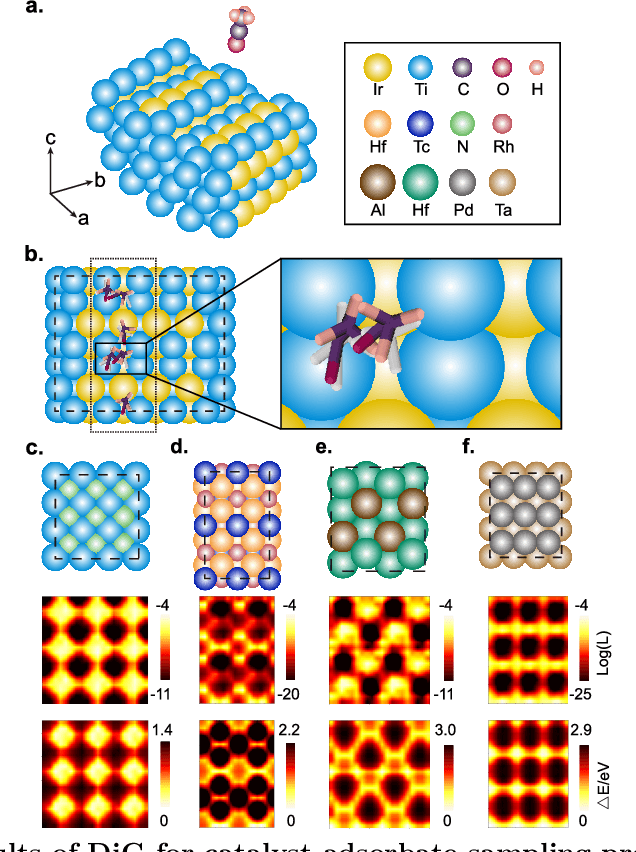
Advances in deep learning have greatly improved structure prediction of molecules. However, many macroscopic observations that are important for real-world applications are not functions of a single molecular structure, but rather determined from the equilibrium distribution of structures. Traditional methods for obtaining these distributions, such as molecular dynamics simulation, are computationally expensive and often intractable. In this paper, we introduce a novel deep learning framework, called Distributional Graphormer (DiG), in an attempt to predict the equilibrium distribution of molecular systems. Inspired by the annealing process in thermodynamics, DiG employs deep neural networks to transform a simple distribution towards the equilibrium distribution, conditioned on a descriptor of a molecular system, such as a chemical graph or a protein sequence. This framework enables efficient generation of diverse conformations and provides estimations of state densities. We demonstrate the performance of DiG on several molecular tasks, including protein conformation sampling, ligand structure sampling, catalyst-adsorbate sampling, and property-guided structure generation. DiG presents a significant advancement in methodology for statistically understanding molecular systems, opening up new research opportunities in molecular science.