 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTurbo2K: Towards Ultra-Efficient and High-Quality 2K Video Synthesis
Apr 20, 2025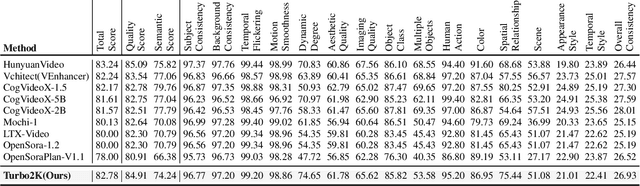

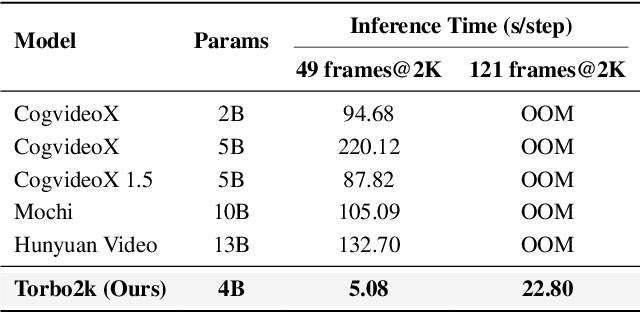

Demand for 2K video synthesis is rising with increasing consumer expectations for ultra-clear visuals. While diffusion transformers (DiTs) have demonstrated remarkable capabilities in high-quality video generation, scaling them to 2K resolution remains computationally prohibitive due to quadratic growth in memory and processing costs. In this work, we propose Turbo2K, an efficient and practical framework for generating detail-rich 2K videos while significantly improving training and inference efficiency. First, Turbo2K operates in a highly compressed latent space, reducing computational complexity and memory footprint, making high-resolution video synthesis feasible. However, the high compression ratio of the VAE and limited model size impose constraints on generative quality. To mitigate this, we introduce a knowledge distillation strategy that enables a smaller student model to inherit the generative capacity of a larger, more powerful teacher model. Our analysis reveals that, despite differences in latent spaces and architectures, DiTs exhibit structural similarities in their internal representations, facilitating effective knowledge transfer. Second, we design a hierarchical two-stage synthesis framework that first generates multi-level feature at lower resolutions before guiding high-resolution video generation. This approach ensures structural coherence and fine-grained detail refinement while eliminating redundant encoding-decoding overhead, further enhancing computational efficiency.Turbo2K achieves state-of-the-art efficiency, generating 5-second, 24fps, 2K videos with significantly reduced computational cost. Compared to existing methods, Turbo2K is up to 20$\times$ faster for inference, making high-resolution video generation more scalable and practical for real-world applications.
Enhancing the Scalability and Applicability of Kohn-Sham Hamiltonians for Molecular Systems
Feb 26, 2025Density Functional Theory (DFT) is a pivotal method within quantum chemistry and materials science, with its core involving the construction and solution of the Kohn-Sham Hamiltonian. Despite its importance, the application of DFT is frequently limited by the substantial computational resources required to construct the Kohn-Sham Hamiltonian. In response to these limitations, current research has employed deep-learning models to efficiently predict molecular and solid Hamiltonians, with roto-translational symmetries encoded in their neural networks. However, the scalability of prior models may be problematic when applied to large molecules, resulting in non-physical predictions of ground-state properties. In this study, we generate a substantially larger training set (PubChemQH) than used previously and use it to create a scalable model for DFT calculations with physical accuracy. For our model, we introduce a loss function derived from physical principles, which we call Wavefunction Alignment Loss (WALoss). WALoss involves performing a basis change on the predicted Hamiltonian to align it with the observed one; thus, the resulting differences can serve as a surrogate for orbital energy differences, allowing models to make better predictions for molecular orbitals and total energies than previously possible. WALoss also substantially accelerates self-consistent-field (SCF) DFT calculations. Here, we show it achieves a reduction in total energy prediction error by a factor of 1347 and an SCF calculation speed-up by a factor of 18%. These substantial improvements set new benchmarks for achieving accurate and applicable predictions in larger molecular systems.
Efficient and Scalable Density Functional Theory Hamiltonian Prediction through Adaptive Sparsity
Feb 03, 2025Hamiltonian matrix prediction is pivotal in computational chemistry, serving as the foundation for determining a wide range of molecular properties. While SE(3) equivariant graph neural networks have achieved remarkable success in this domain, their substantial computational cost-driven by high-order tensor product (TP) operations-restricts their scalability to large molecular systems with extensive basis sets. To address this challenge, we introduce SPHNet, an efficient and scalable equivariant network that incorporates adaptive sparsity into Hamiltonian prediction. SPHNet employs two innovative sparse gates to selectively constrain non-critical interaction combinations, significantly reducing tensor product computations while maintaining accuracy. To optimize the sparse representation, we develop a Three-phase Sparsity Scheduler, ensuring stable convergence and achieving high performance at sparsity rates of up to 70 percent. Extensive evaluations on QH9 and PubchemQH datasets demonstrate that SPHNet achieves state-of-the-art accuracy while providing up to a 7x speedup over existing models. Beyond Hamiltonian prediction, the proposed sparsification techniques also hold significant potential for improving the efficiency and scalability of other SE(3) equivariant networks, further broadening their applicability and impact.
MagicEraser: Erasing Any Objects via Semantics-Aware Control
Oct 14, 2024The traditional image inpainting task aims to restore corrupted regions by referencing surrounding background and foreground. However, the object erasure task, which is in increasing demand, aims to erase objects and generate harmonious background. Previous GAN-based inpainting methods struggle with intricate texture generation. Emerging diffusion model-based algorithms, such as Stable Diffusion Inpainting, exhibit the capability to generate novel content, but they often produce incongruent results at the locations of the erased objects and require high-quality text prompt inputs. To address these challenges, we introduce MagicEraser, a diffusion model-based framework tailored for the object erasure task. It consists of two phases: content initialization and controllable generation. In the latter phase, we develop two plug-and-play modules called prompt tuning and semantics-aware attention refocus. Additionally, we propose a data construction strategy that generates training data specially suitable for this task. MagicEraser achieves fine and effective control of content generation while mitigating undesired artifacts. Experimental results highlight a valuable advancement of our approach in the object erasure task.
Neural P$^3$M: A Long-Range Interaction Modeling Enhancer for Geometric GNNs
Sep 26, 2024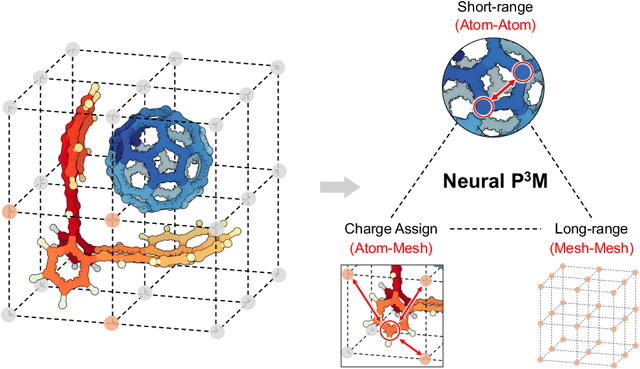
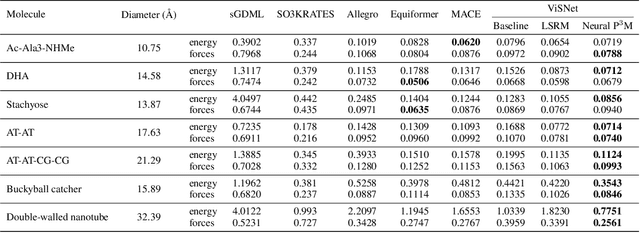
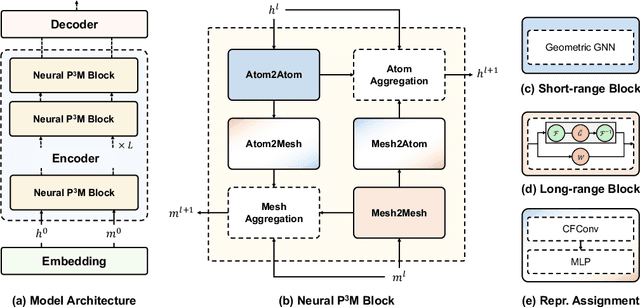
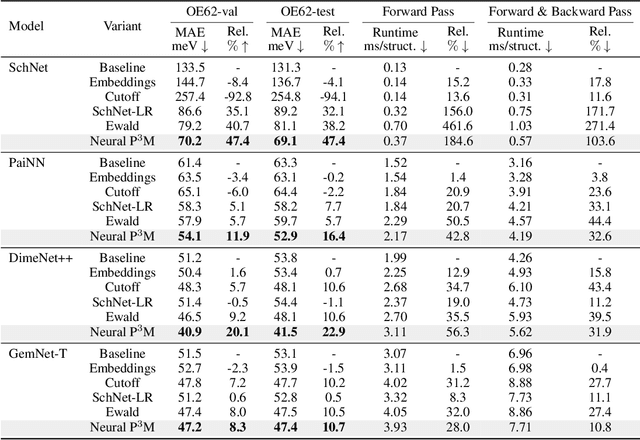
Geometric graph neural networks (GNNs) have emerged as powerful tools for modeling molecular geometry. However, they encounter limitations in effectively capturing long-range interactions in large molecular systems. To address this challenge, we introduce Neural P$^3$M, a versatile enhancer of geometric GNNs to expand the scope of their capabilities by incorporating mesh points alongside atoms and reimaging traditional mathematical operations in a trainable manner. Neural P$^3$M exhibits flexibility across a wide range of molecular systems and demonstrates remarkable accuracy in predicting energies and forces, outperforming on benchmarks such as the MD22 dataset. It also achieves an average improvement of 22% on the OE62 dataset while integrating with various architectures.
UltraPixel: Advancing Ultra-High-Resolution Image Synthesis to New Peaks
Jul 02, 2024



Ultra-high-resolution image generation poses great challenges, such as increased semantic planning complexity and detail synthesis difficulties, alongside substantial training resource demands. We present UltraPixel, a novel architecture utilizing cascade diffusion models to generate high-quality images at multiple resolutions (\textit{e.g.}, 1K to 6K) within a single model, while maintaining computational efficiency. UltraPixel leverages semantics-rich representations of lower-resolution images in the later denoising stage to guide the whole generation of highly detailed high-resolution images, significantly reducing complexity. Furthermore, we introduce implicit neural representations for continuous upsampling and scale-aware normalization layers adaptable to various resolutions. Notably, both low- and high-resolution processes are performed in the most compact space, sharing the majority of parameters with less than 3$\%$ additional parameters for high-resolution outputs, largely enhancing training and inference efficiency. Our model achieves fast training with reduced data requirements, producing photo-realistic high-resolution images and demonstrating state-of-the-art performance in extensive experiments.
Infusing Self-Consistency into Density Functional Theory Hamiltonian Prediction via Deep Equilibrium Models
Jun 06, 2024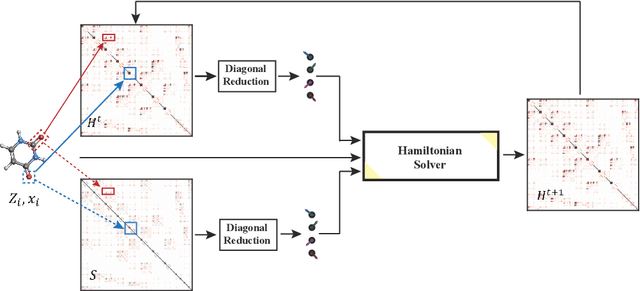
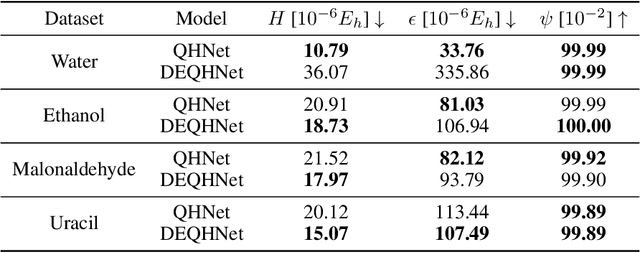
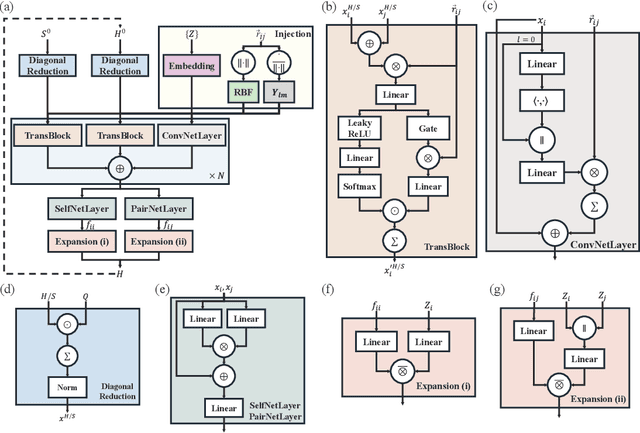
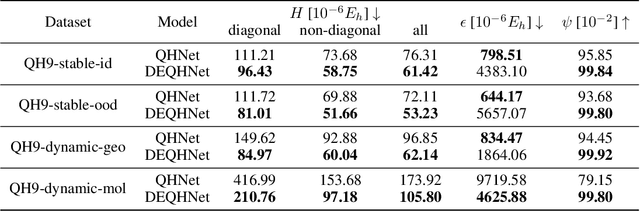
In this study, we introduce a unified neural network architecture, the Deep Equilibrium Density Functional Theory Hamiltonian (DEQH) model, which incorporates Deep Equilibrium Models (DEQs) for predicting Density Functional Theory (DFT) Hamiltonians. The DEQH model inherently captures the self-consistency nature of Hamiltonian, a critical aspect often overlooked by traditional machine learning approaches for Hamiltonian prediction. By employing DEQ within our model architecture, we circumvent the need for DFT calculations during the training phase to introduce the Hamiltonian's self-consistency, thus addressing computational bottlenecks associated with large or complex systems. We propose a versatile framework that combines DEQ with off-the-shelf machine learning models for predicting Hamiltonians. When benchmarked on the MD17 and QH9 datasets, DEQHNet, an instantiation of the DEQH framework, has demonstrated a significant improvement in prediction accuracy. Beyond a predictor, the DEQH model is a Hamiltonian solver, in the sense that it uses the fixed-point solving capability of the deep equilibrium model to iteratively solve for the Hamiltonian. Ablation studies of DEQHNet further elucidate the network's effectiveness, offering insights into the potential of DEQ-integrated networks for Hamiltonian learning.
SE3Set: Harnessing equivariant hypergraph neural networks for molecular representation learning
May 26, 2024In this paper, we develop SE3Set, an SE(3) equivariant hypergraph neural network architecture tailored for advanced molecular representation learning. Hypergraphs are not merely an extension of traditional graphs; they are pivotal for modeling high-order relationships, a capability that conventional equivariant graph-based methods lack due to their inherent limitations in representing intricate many-body interactions. To achieve this, we first construct hypergraphs via proposing a new fragmentation method that considers both chemical and three-dimensional spatial information of molecular system. We then design SE3Set, which incorporates equivariance into the hypergragh neural network. This ensures that the learned molecular representations are invariant to spatial transformations, thereby providing robustness essential for accurate prediction of molecular properties. SE3Set has shown performance on par with state-of-the-art (SOTA) models for small molecule datasets like QM9 and MD17. It excels on the MD22 dataset, achieving a notable improvement of approximately 20% in accuracy across all molecules, which highlights the prevalence of complex many-body interactions in larger molecules. This exceptional performance of SE3Set across diverse molecular structures underscores its transformative potential in computational chemistry, offering a route to more accurate and physically nuanced modeling.
F$^3$low: Frame-to-Frame Coarse-grained Molecular Dynamics with SE Guided Flow Matching
May 01, 2024



Molecular dynamics (MD) is a crucial technique for simulating biological systems, enabling the exploration of their dynamic nature and fostering an understanding of their functions and properties. To address exploration inefficiency, emerging enhanced sampling approaches like coarse-graining (CG) and generative models have been employed. In this work, we propose a \underline{Frame-to-Frame} generative model with guided \underline{Flow}-matching (F$3$low) for enhanced sampling, which (a) extends the domain of CG modeling to the SE(3) Riemannian manifold; (b) retreating CGMD simulations as autoregressively sampling guided by the former frame via flow-matching models; (c) targets the protein backbone, offering improved insights into secondary structure formation and intricate folding pathways. Compared to previous methods, F$3$low allows for broader exploration of conformational space. The ability to rapidly generate diverse conformations via force-free generative paradigm on SE(3) paves the way toward efficient enhanced sampling methods.
Self-Consistency Training for Hamiltonian Prediction
Mar 14, 2024



Hamiltonian prediction is a versatile formulation to leverage machine learning for solving molecular science problems. Yet, its applicability is limited by insufficient labeled data for training. In this work, we highlight that Hamiltonian prediction possesses a self-consistency principle, based on which we propose an exact training method that does not require labeled data. This merit addresses the data scarcity difficulty, and distinguishes the task from other property prediction formulations with unique benefits: (1) self-consistency training enables the model to be trained on a large amount of unlabeled data, hence substantially enhances generalization; (2) self-consistency training is more efficient than labeling data with DFT for supervised training, since it is an amortization of DFT calculation over a set of molecular structures. We empirically demonstrate the better generalization in data-scarce and out-of-distribution scenarios, and the better efficiency from the amortization. These benefits push forward the applicability of Hamiltonian prediction to an ever larger scale.