 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNeural P$^3$M: A Long-Range Interaction Modeling Enhancer for Geometric GNNs
Sep 26, 2024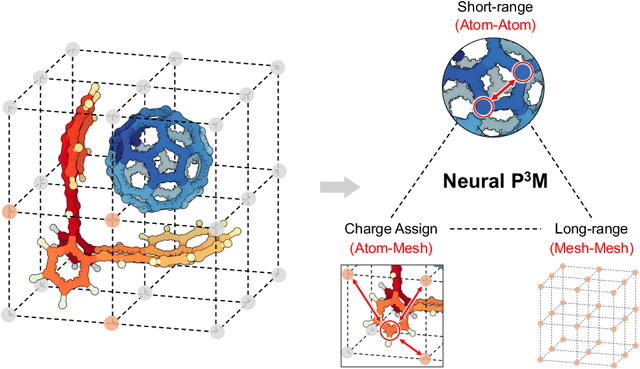
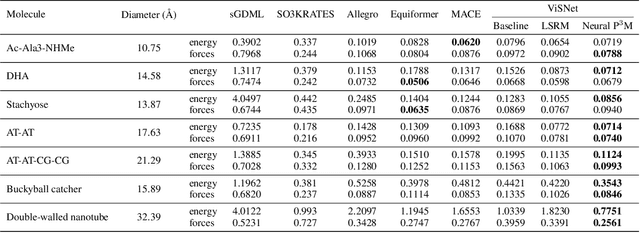
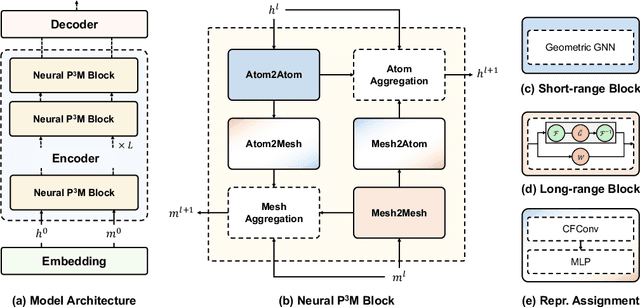
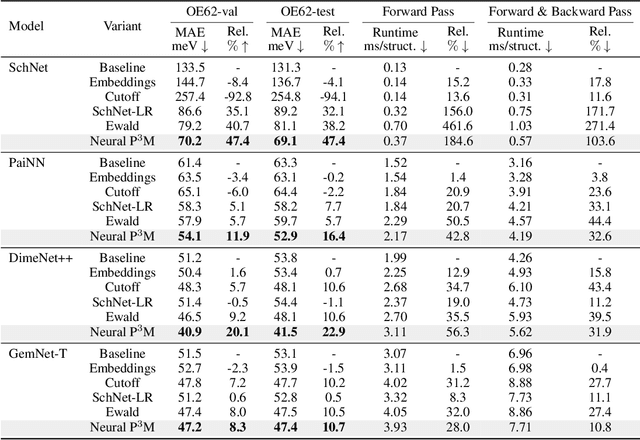
Geometric graph neural networks (GNNs) have emerged as powerful tools for modeling molecular geometry. However, they encounter limitations in effectively capturing long-range interactions in large molecular systems. To address this challenge, we introduce Neural P$^3$M, a versatile enhancer of geometric GNNs to expand the scope of their capabilities by incorporating mesh points alongside atoms and reimaging traditional mathematical operations in a trainable manner. Neural P$^3$M exhibits flexibility across a wide range of molecular systems and demonstrates remarkable accuracy in predicting energies and forces, outperforming on benchmarks such as the MD22 dataset. It also achieves an average improvement of 22% on the OE62 dataset while integrating with various architectures.
F$^3$low: Frame-to-Frame Coarse-grained Molecular Dynamics with SE Guided Flow Matching
May 01, 2024



Molecular dynamics (MD) is a crucial technique for simulating biological systems, enabling the exploration of their dynamic nature and fostering an understanding of their functions and properties. To address exploration inefficiency, emerging enhanced sampling approaches like coarse-graining (CG) and generative models have been employed. In this work, we propose a \underline{Frame-to-Frame} generative model with guided \underline{Flow}-matching (F$3$low) for enhanced sampling, which (a) extends the domain of CG modeling to the SE(3) Riemannian manifold; (b) retreating CGMD simulations as autoregressively sampling guided by the former frame via flow-matching models; (c) targets the protein backbone, offering improved insights into secondary structure formation and intricate folding pathways. Compared to previous methods, F$3$low allows for broader exploration of conformational space. The ability to rapidly generate diverse conformations via force-free generative paradigm on SE(3) paves the way toward efficient enhanced sampling methods.
An ensemble of VisNet, Transformer-M, and pretraining models for molecular property prediction in OGB Large-Scale Challenge @ NeurIPS 2022
Nov 23, 2022



In the technical report, we provide our solution for OGB-LSC 2022 Graph Regression Task. The target of this task is to predict the quantum chemical property, HOMO-LUMO gap for a given molecule on PCQM4Mv2 dataset. In the competition, we designed two kinds of models: Transformer-M-ViSNet which is an geometry-enhanced graph neural network for fully connected molecular graphs and Pretrained-3D-ViSNet which is a pretrained ViSNet by distilling geomeotric information from optimized structures. With an ensemble of 22 models, ViSNet Team achieved the MAE of 0.0723 eV on the test-challenge set, dramatically reducing the error by 39.75% compared with the best method in the last year competition.