 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGPCR-Filter: a deep learning framework for efficient and precise GPCR modulator discovery
Jan 27, 2026G protein-coupled receptors (GPCRs) govern diverse physiological processes and are central to modern pharmacology. Yet discovering GPCR modulators remains challenging because receptor activation often arises from complex allosteric effects rather than direct binding affinity, and conventional assays are slow, costly, and not optimized for capturing these dynamics. Here we present GPCR-Filter, a deep learning framework specifically developed for GPCR modulator discovery. We assembled a high-quality dataset of over 90,000 experimentally validated GPCR-ligand pairs, providing a robust foundation for training and evaluation. GPCR-Filter integrates the ESM-3 protein language model for high-fidelity GPCR sequence representations with graph neural networks that encode ligand structures, coupled through an attention-based fusion mechanism that learns receptor-ligand functional relationships. Across multiple evaluation settings, GPCR-Filter consistently outperforms state-of-the-art compound-protein interaction models and exhibits strong generalization to unseen receptors and ligands. Notably, the model successfully identified micromolar-level agonists of the 5-HT\textsubscript{1A} receptor with distinct chemical frameworks. These results establish GPCR-Filter as a scalable and effective computational approach for GPCR modulator discovery, advancing AI-assisted drug development for complex signaling systems.
Peptide2Mol: A Diffusion Model for Generating Small Molecules as Peptide Mimics for Targeted Protein Binding
Nov 07, 2025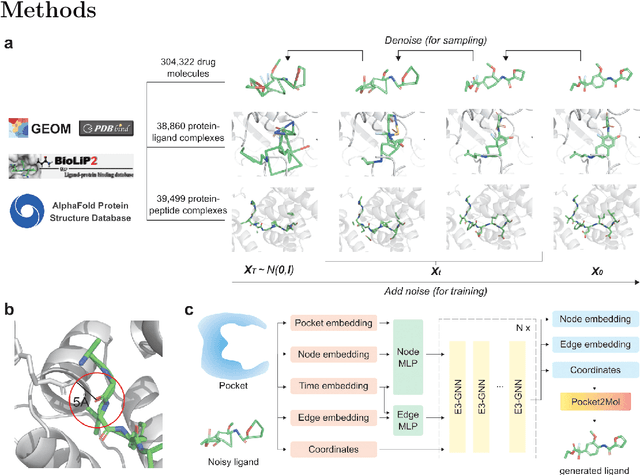
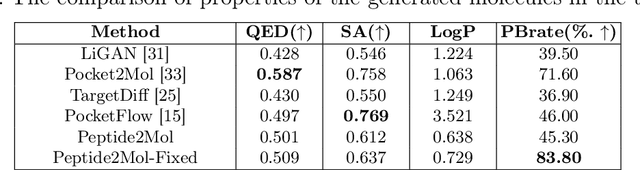
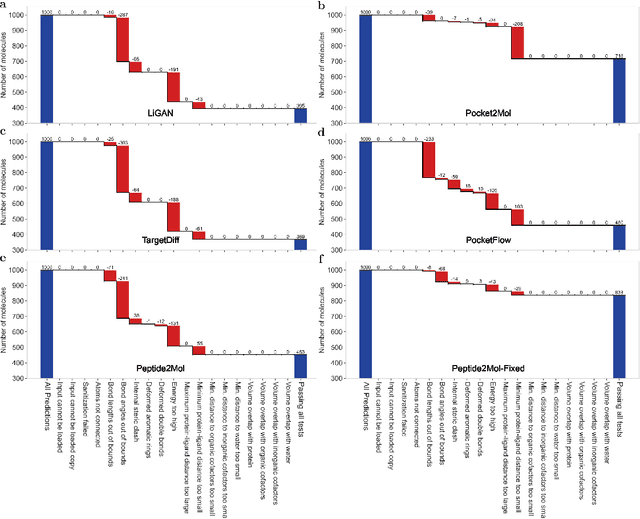
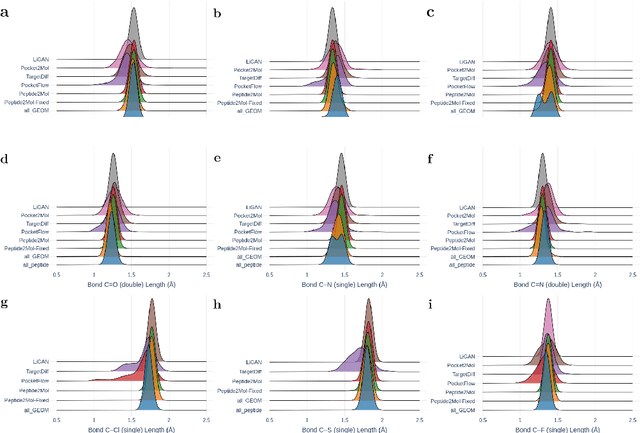
Structure-based drug design has seen significant advancements with the integration of artificial intelligence (AI), particularly in the generation of hit and lead compounds. However, most AI-driven approaches neglect the importance of endogenous protein interactions with peptides, which may result in suboptimal molecule designs. In this work, we present Peptide2Mol, an E(3)-equivariant graph neural network diffusion model that generates small molecules by referencing both the original peptide binders and their surrounding protein pocket environments. Trained on large datasets and leveraging sophisticated modeling techniques, Peptide2Mol not only achieves state-of-the-art performance in non-autoregressive generative tasks, but also produces molecules with similarity to the original peptide binder. Additionally, the model allows for molecule optimization and peptidomimetic design through a partial diffusion process. Our results highlight Peptide2Mol as an effective deep generative model for generating and optimizing bioactive small molecules from protein binding pockets.
Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows
Mar 06, 2025The dynamic nature of proteins, influenced by ligand interactions, is essential for comprehending protein function and progressing drug discovery. Traditional structure-based drug design (SBDD) approaches typically target binding sites with rigid structures, limiting their practical application in drug development. While molecular dynamics simulation can theoretically capture all the biologically relevant conformations, the transition rate is dictated by the intrinsic energy barrier between them, making the sampling process computationally expensive. To overcome the aforementioned challenges, we propose to use generative modeling for SBDD considering conformational changes of protein pockets. We curate a dataset of apo and multiple holo states of protein-ligand complexes, simulated by molecular dynamics, and propose a full-atom flow model (and a stochastic version), named DynamicFlow, that learns to transform apo pockets and noisy ligands into holo pockets and corresponding 3D ligand molecules. Our method uncovers promising ligand molecules and corresponding holo conformations of pockets. Additionally, the resultant holo-like states provide superior inputs for traditional SBDD approaches, playing a significant role in practical drug discovery.
An ensemble of VisNet, Transformer-M, and pretraining models for molecular property prediction in OGB Large-Scale Challenge @ NeurIPS 2022
Nov 23, 2022



In the technical report, we provide our solution for OGB-LSC 2022 Graph Regression Task. The target of this task is to predict the quantum chemical property, HOMO-LUMO gap for a given molecule on PCQM4Mv2 dataset. In the competition, we designed two kinds of models: Transformer-M-ViSNet which is an geometry-enhanced graph neural network for fully connected molecular graphs and Pretrained-3D-ViSNet which is a pretrained ViSNet by distilling geomeotric information from optimized structures. With an ensemble of 22 models, ViSNet Team achieved the MAE of 0.0723 eV on the test-challenge set, dramatically reducing the error by 39.75% compared with the best method in the last year competition.