 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgePeptide2Mol: A Diffusion Model for Generating Small Molecules as Peptide Mimics for Targeted Protein Binding
Nov 07, 2025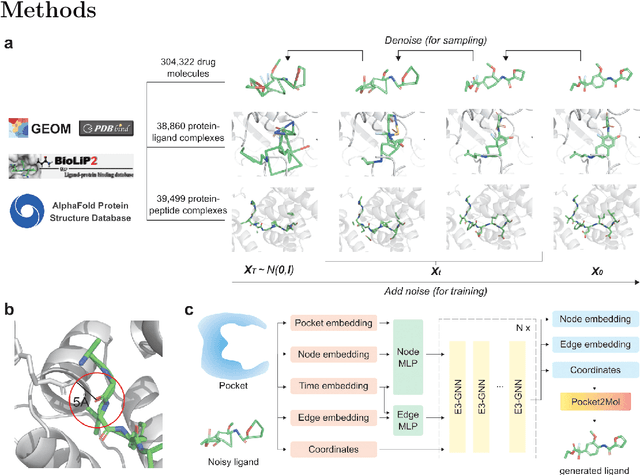
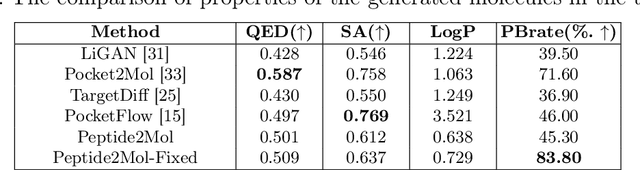
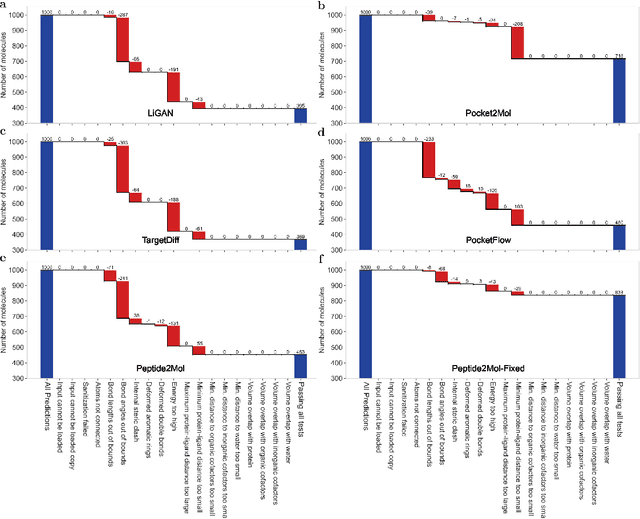
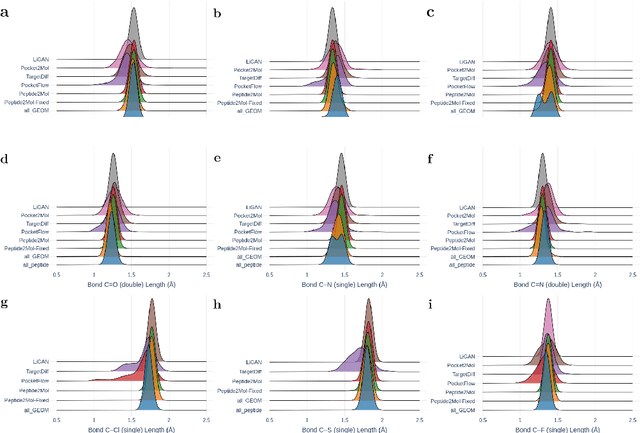
Structure-based drug design has seen significant advancements with the integration of artificial intelligence (AI), particularly in the generation of hit and lead compounds. However, most AI-driven approaches neglect the importance of endogenous protein interactions with peptides, which may result in suboptimal molecule designs. In this work, we present Peptide2Mol, an E(3)-equivariant graph neural network diffusion model that generates small molecules by referencing both the original peptide binders and their surrounding protein pocket environments. Trained on large datasets and leveraging sophisticated modeling techniques, Peptide2Mol not only achieves state-of-the-art performance in non-autoregressive generative tasks, but also produces molecules with similarity to the original peptide binder. Additionally, the model allows for molecule optimization and peptidomimetic design through a partial diffusion process. Our results highlight Peptide2Mol as an effective deep generative model for generating and optimizing bioactive small molecules from protein binding pockets.
Morpheus: A Neural-driven Animatronic Face with Hybrid Actuation and Diverse Emotion Control
Jul 22, 2025Previous animatronic faces struggle to express emotions effectively due to hardware and software limitations. On the hardware side, earlier approaches either use rigid-driven mechanisms, which provide precise control but are difficult to design within constrained spaces, or tendon-driven mechanisms, which are more space-efficient but challenging to control. In contrast, we propose a hybrid actuation approach that combines the best of both worlds. The eyes and mouth-key areas for emotional expression-are controlled using rigid mechanisms for precise movement, while the nose and cheek, which convey subtle facial microexpressions, are driven by strings. This design allows us to build a compact yet versatile hardware platform capable of expressing a wide range of emotions. On the algorithmic side, our method introduces a self-modeling network that maps motor actions to facial landmarks, allowing us to automatically establish the relationship between blendshape coefficients for different facial expressions and the corresponding motor control signals through gradient backpropagation. We then train a neural network to map speech input to corresponding blendshape controls. With our method, we can generate distinct emotional expressions such as happiness, fear, disgust, and anger, from any given sentence, each with nuanced, emotion-specific control signals-a feature that has not been demonstrated in earlier systems. We release the hardware design and code at https://github.com/ZZongzheng0918/Morpheus-Hardware and https://github.com/ZZongzheng0918/Morpheus-Software.
Inference-time Scaling of Diffusion Models through Classical Search
May 29, 2025Classical search algorithms have long underpinned modern artificial intelligence. In this work, we tackle the challenge of inference-time control in diffusion models -- adapting generated outputs to meet diverse test-time objectives -- using principles from classical search. We propose a general framework that orchestrates local and global search to efficiently navigate the generative space. It employs a theoretically grounded local search via annealed Langevin MCMC and performs compute-efficient global exploration using breadth-first and depth-first tree search. We evaluate our approach on a range of challenging domains, including planning, offline reinforcement learning, and image generation. Across all tasks, we observe significant gains in both performance and efficiency. These results show that classical search provides a principled and practical foundation for inference-time scaling in diffusion models. Project page at diffusion-inference-scaling.github.io.
Designing Cyclic Peptides via Harmonic SDE with Atom-Bond Modeling
May 27, 2025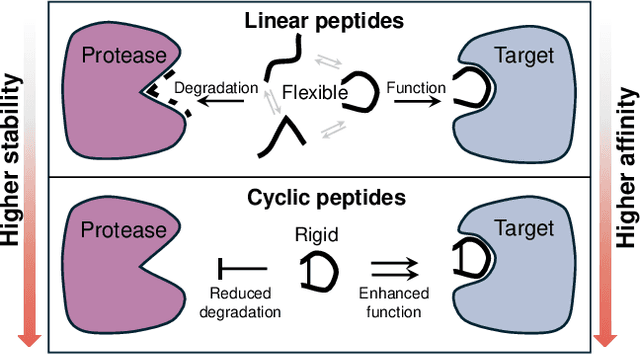
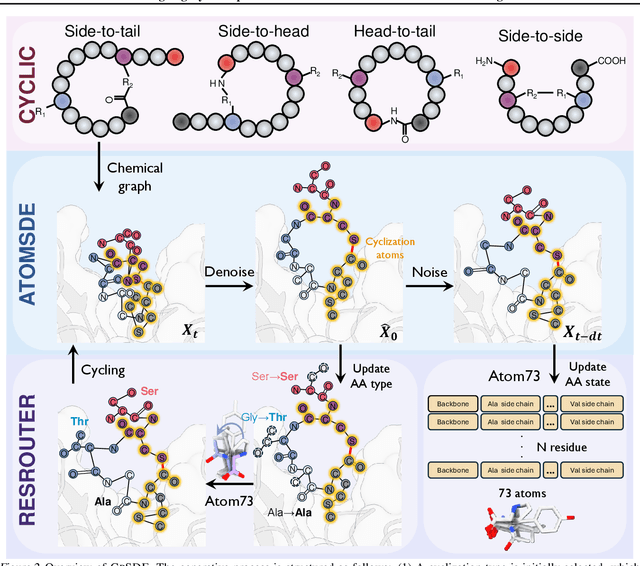
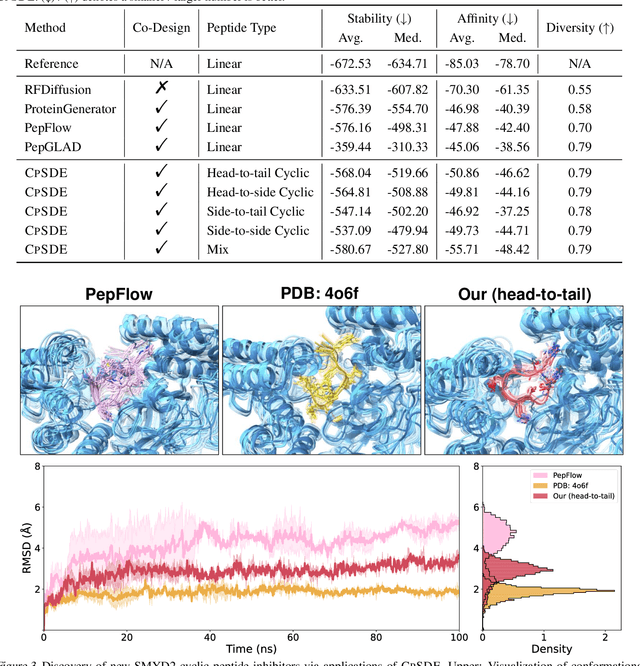
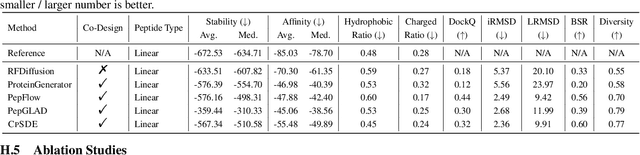
Cyclic peptides offer inherent advantages in pharmaceuticals. For example, cyclic peptides are more resistant to enzymatic hydrolysis compared to linear peptides and usually exhibit excellent stability and affinity. Although deep generative models have achieved great success in linear peptide design, several challenges prevent the development of computational methods for designing diverse types of cyclic peptides. These challenges include the scarcity of 3D structural data on target proteins and associated cyclic peptide ligands, the geometric constraints that cyclization imposes, and the involvement of non-canonical amino acids in cyclization. To address the above challenges, we introduce CpSDE, which consists of two key components: AtomSDE, a generative structure prediction model based on harmonic SDE, and ResRouter, a residue type predictor. Utilizing a routed sampling algorithm that alternates between these two models to iteratively update sequences and structures, CpSDE facilitates the generation of cyclic peptides. By employing explicit all-atom and bond modeling, CpSDE overcomes existing data limitations and is proficient in designing a wide variety of cyclic peptides. Our experimental results demonstrate that the cyclic peptides designed by our method exhibit reliable stability and affinity.
Generative Evaluation of Complex Reasoning in Large Language Models
Apr 03, 2025
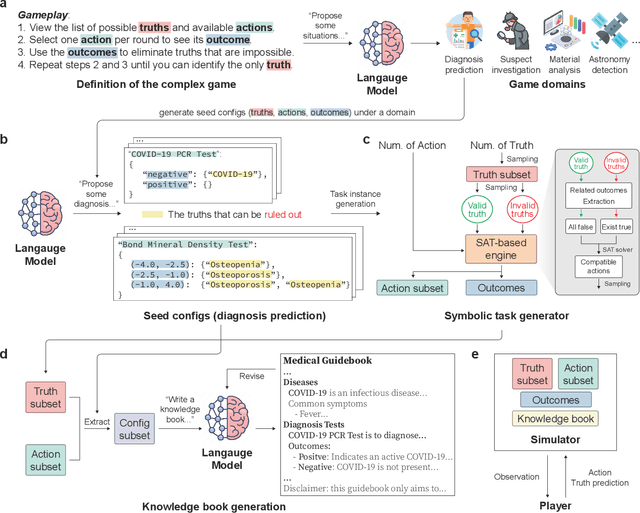
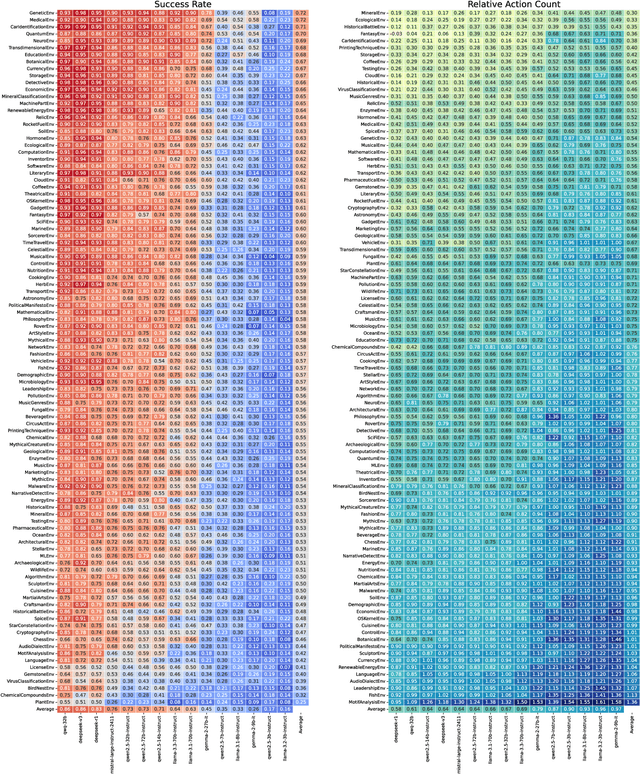
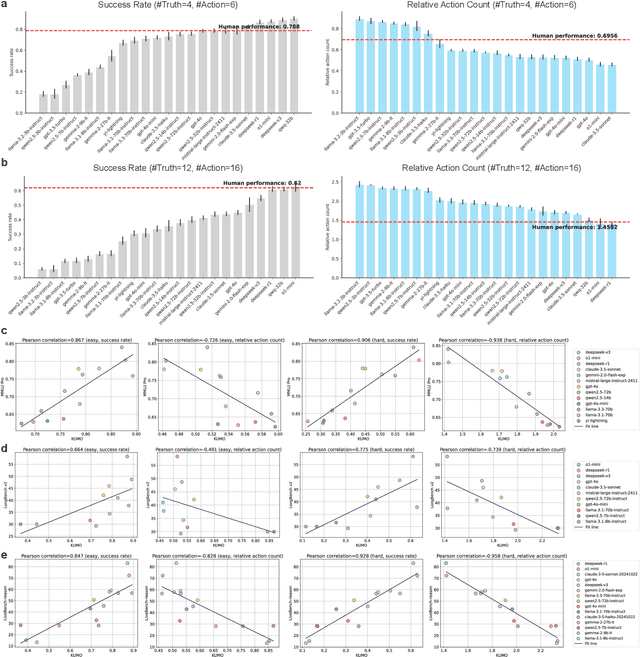
With powerful large language models (LLMs) demonstrating superhuman reasoning capabilities, a critical question arises: Do LLMs genuinely reason, or do they merely recall answers from their extensive, web-scraped training datasets? Publicly released benchmarks inevitably become contaminated once incorporated into subsequent LLM training sets, undermining their reliability as faithful assessments. To address this, we introduce KUMO, a generative evaluation framework designed specifically for assessing reasoning in LLMs. KUMO synergistically combines LLMs with symbolic engines to dynamically produce diverse, multi-turn reasoning tasks that are partially observable and adjustable in difficulty. Through an automated pipeline, KUMO continuously generates novel tasks across open-ended domains, compelling models to demonstrate genuine generalization rather than memorization. We evaluated 23 state-of-the-art LLMs on 5,000 tasks across 100 domains created by KUMO, benchmarking their reasoning abilities against university students. Our findings reveal that many LLMs have outperformed university-level performance on easy reasoning tasks, and reasoning-scaled LLMs reach university-level performance on complex reasoning challenges. Moreover, LLM performance on KUMO tasks correlates strongly with results on newly released real-world reasoning benchmarks, underscoring KUMO's value as a robust, enduring assessment tool for genuine LLM reasoning capabilities.
UniMoMo: Unified Generative Modeling of 3D Molecules for De Novo Binder Design
Mar 25, 2025The design of target-specific molecules such as small molecules, peptides, and antibodies is vital for biological research and drug discovery. Existing generative methods are restricted to single-domain molecules, failing to address versatile therapeutic needs or utilize cross-domain transferability to enhance model performance. In this paper, we introduce Unified generative Modeling of 3D Molecules (UniMoMo), the first framework capable of designing binders of multiple molecular domains using a single model. In particular, UniMoMo unifies the representations of different molecules as graphs of blocks, where each block corresponds to either a standard amino acid or a molecular fragment. Based on these unified representations, UniMoMo utilizes a geometric latent diffusion model for 3D molecular generation, featuring an iterative full-atom autoencoder to compress blocks into latent space points, followed by an E(3)-equivariant diffusion process. Extensive benchmarks across peptides, antibodies, and small molecules demonstrate the superiority of our unified framework over existing domain-specific models, highlighting the benefits of multi-domain training.
A Neural Symbolic Model for Space Physics
Mar 11, 2025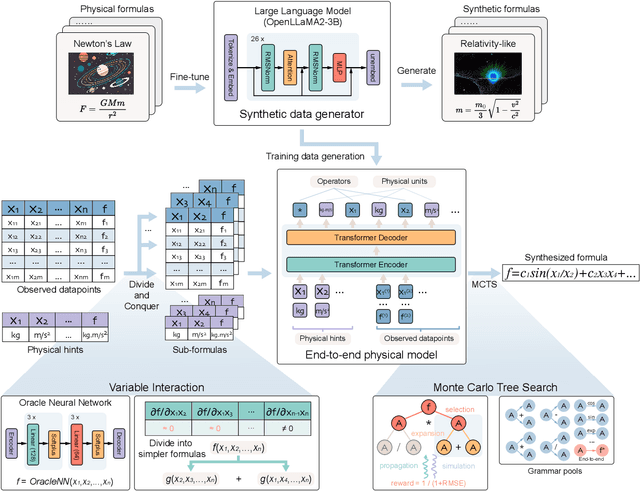
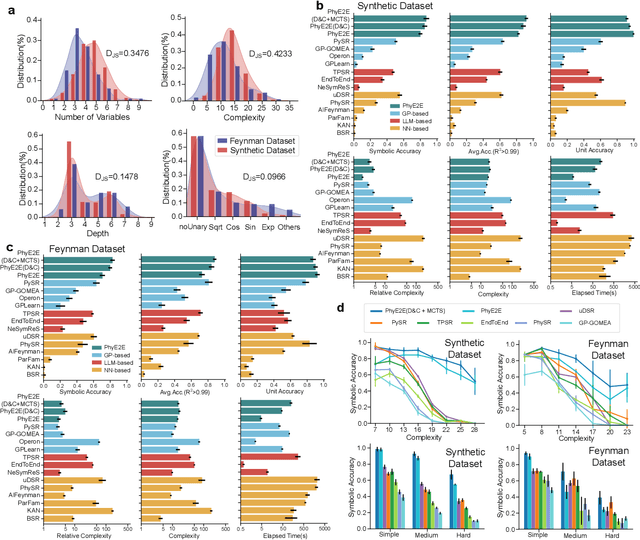
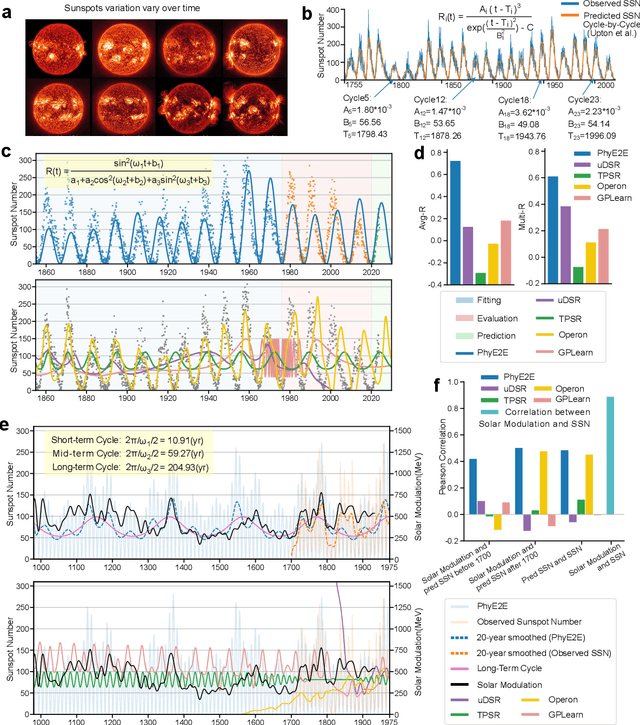
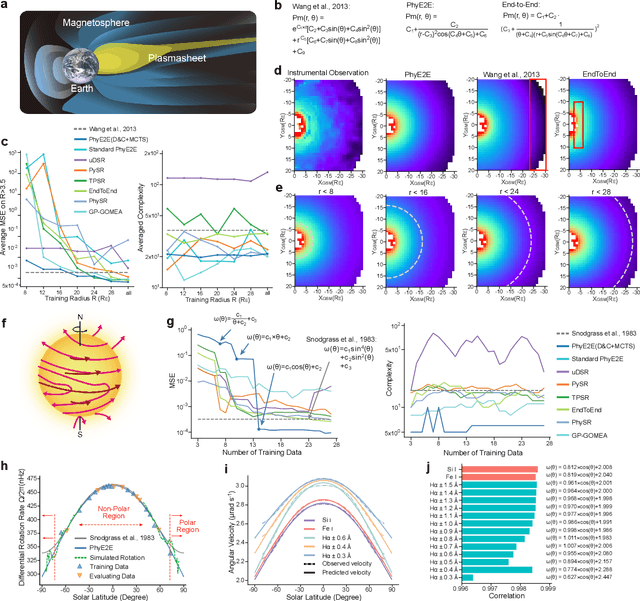
In this study, we unveil a new AI model, termed PhyE2E, to discover physical formulas through symbolic regression. PhyE2E simplifies symbolic regression by decomposing it into sub-problems using the second-order derivatives of an oracle neural network, and employs a transformer model to translate data into symbolic formulas in an end-to-end manner. The resulting formulas are refined through Monte-Carlo Tree Search and Genetic Programming. We leverage a large language model to synthesize extensive symbolic expressions resembling real physics, and train the model to recover these formulas directly from data. A comprehensive evaluation reveals that PhyE2E outperforms existing state-of-the-art approaches, delivering superior symbolic accuracy, precision in data fitting, and consistency in physical units. We deployed PhyE2E to five applications in space physics, including the prediction of sunspot numbers, solar rotational angular velocity, emission line contribution functions, near-Earth plasma pressure, and lunar-tide plasma signals. The physical formulas generated by AI demonstrate a high degree of accuracy in fitting the experimental data from satellites and astronomical telescopes. We have successfully upgraded the formula proposed by NASA in 1993 regarding solar activity, and for the first time, provided the explanations for the long cycle of solar activity in an explicit form. We also found that the decay of near-Earth plasma pressure is proportional to r^2 to Earth, where subsequent mathematical derivations are consistent with satellite data from another independent study. Moreover, we found physical formulas that can describe the relationships between emission lines in the extreme ultraviolet spectrum of the Sun, temperatures, electron densities, and magnetic fields. The formula obtained is consistent with the properties that physicists had previously hypothesized it should possess.
Integrating Protein Dynamics into Structure-Based Drug Design via Full-Atom Stochastic Flows
Mar 06, 2025The dynamic nature of proteins, influenced by ligand interactions, is essential for comprehending protein function and progressing drug discovery. Traditional structure-based drug design (SBDD) approaches typically target binding sites with rigid structures, limiting their practical application in drug development. While molecular dynamics simulation can theoretically capture all the biologically relevant conformations, the transition rate is dictated by the intrinsic energy barrier between them, making the sampling process computationally expensive. To overcome the aforementioned challenges, we propose to use generative modeling for SBDD considering conformational changes of protein pockets. We curate a dataset of apo and multiple holo states of protein-ligand complexes, simulated by molecular dynamics, and propose a full-atom flow model (and a stochastic version), named DynamicFlow, that learns to transform apo pockets and noisy ligands into holo pockets and corresponding 3D ligand molecules. Our method uncovers promising ligand molecules and corresponding holo conformations of pockets. Additionally, the resultant holo-like states provide superior inputs for traditional SBDD approaches, playing a significant role in practical drug discovery.
MatterChat: A Multi-Modal LLM for Material Science
Feb 18, 2025


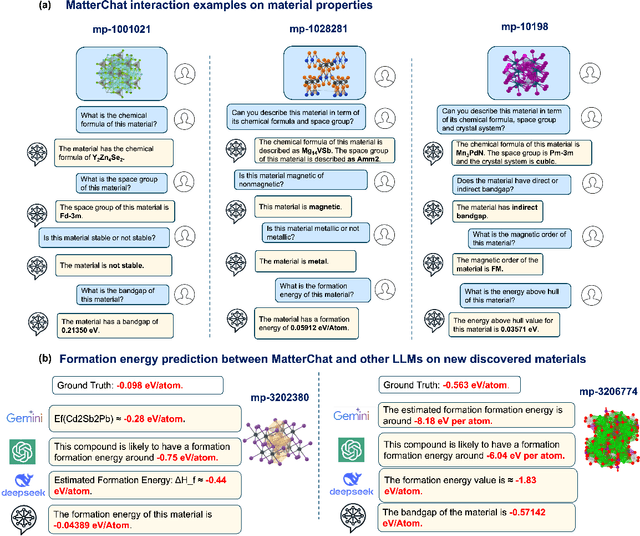
Understanding and predicting the properties of inorganic materials is crucial for accelerating advancements in materials science and driving applications in energy, electronics, and beyond. Integrating material structure data with language-based information through multi-modal large language models (LLMs) offers great potential to support these efforts by enhancing human-AI interaction. However, a key challenge lies in integrating atomic structures at full resolution into LLMs. In this work, we introduce MatterChat, a versatile structure-aware multi-modal LLM that unifies material structural data and textual inputs into a single cohesive model. MatterChat employs a bridging module to effectively align a pretrained machine learning interatomic potential with a pretrained LLM, reducing training costs and enhancing flexibility. Our results demonstrate that MatterChat significantly improves performance in material property prediction and human-AI interaction, surpassing general-purpose LLMs such as GPT-4. We also demonstrate its usefulness in applications such as more advanced scientific reasoning and step-by-step material synthesis.
PUGS: Zero-shot Physical Understanding with Gaussian Splatting
Feb 17, 2025
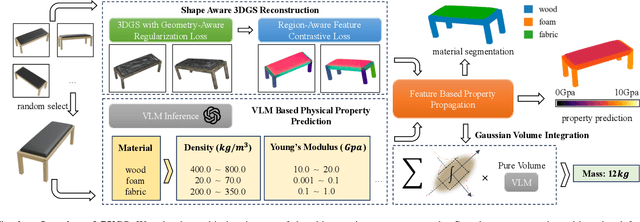

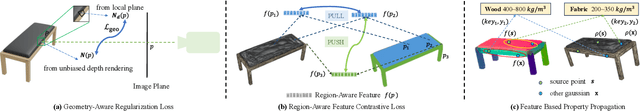
Current robotic systems can understand the categories and poses of objects well. But understanding physical properties like mass, friction, and hardness, in the wild, remains challenging. We propose a new method that reconstructs 3D objects using the Gaussian splatting representation and predicts various physical properties in a zero-shot manner. We propose two techniques during the reconstruction phase: a geometry-aware regularization loss function to improve the shape quality and a region-aware feature contrastive loss function to promote region affinity. Two other new techniques are designed during inference: a feature-based property propagation module and a volume integration module tailored for the Gaussian representation. Our framework is named as zero-shot physical understanding with Gaussian splatting, or PUGS. PUGS achieves new state-of-the-art results on the standard benchmark of ABO-500 mass prediction. We provide extensive quantitative ablations and qualitative visualization to demonstrate the mechanism of our designs. We show the proposed methodology can help address challenging real-world grasping tasks. Our codes, data, and models are available at https://github.com/EverNorif/PUGS