 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDMFlow: Disordered Materials Generation by Flow Matching
Feb 04, 2026The design of materials with tailored properties is crucial for technological progress. However, most deep generative models focus exclusively on perfectly ordered crystals, neglecting the important class of disordered materials. To address this gap, we introduce DMFlow, a generative framework specifically designed for disordered crystals. Our approach introduces a unified representation for ordered, Substitutionally Disordered (SD), and Positionally Disordered (PD) crystals, and employs a flow matching model to jointly generate all structural components. A key innovation is a Riemannian flow matching framework with spherical reparameterization, which ensures physically valid disorder weights on the probability simplex. The vector field is learned by a novel Graph Neural Network (GNN) that incorporates physical symmetries and a specialized message-passing scheme. Finally, a two-stage discretization procedure converts the continuous weights into multi-hot atomic assignments. To support research in this area, we release a benchmark containing SD, PD, and mixed structures curated from the Crystallography Open Database. Experiments on Crystal Structure Prediction (CSP) and De Novo Generation (DNG) tasks demonstrate that DMFlow significantly outperforms state-of-the-art baselines adapted from ordered crystal generation. We hope our work provides a foundation for the AI-driven discovery of disordered materials.
Optimization and Generation in Aerodynamics Inverse Design
Feb 03, 2026Inverse design with physics-based objectives is challenging because it couples high-dimensional geometry with expensive simulations, as exemplified by aerodynamic shape optimization for drag reduction. We revisit inverse design through two canonical solutions, the optimal design point and the optimal design distribution, and relate them to optimization and guided generation. Building on this view, we propose a new training loss for cost predictors and a density-gradient optimization method that improves objectives while preserving plausible shapes. We further unify existing training-free guided generation methods. To address their inability to approximate conditional covariance in high dimensions, we develop a time- and memory-efficient algorithm for approximate covariance estimation. Experiments on a controlled 2D study and high-fidelity 3D aerodynamic benchmarks (car and aircraft), validated by OpenFOAM simulations and miniature wind-tunnel tests with 3D-printed prototypes, demonstrate consistent gains in both optimization and guided generation. Additional offline RL results further support the generality of our approach.
CloDS: Visual-Only Unsupervised Cloth Dynamics Learning in Unknown Conditions
Feb 02, 2026Deep learning has demonstrated remarkable capabilities in simulating complex dynamic systems. However, existing methods require known physical properties as supervision or inputs, limiting their applicability under unknown conditions. To explore this challenge, we introduce Cloth Dynamics Grounding (CDG), a novel scenario for unsupervised learning of cloth dynamics from multi-view visual observations. We further propose Cloth Dynamics Splatting (CloDS), an unsupervised dynamic learning framework designed for CDG. CloDS adopts a three-stage pipeline that first performs video-to-geometry grounding and then trains a dynamics model on the grounded meshes. To cope with large non-linear deformations and severe self-occlusions during grounding, we introduce a dual-position opacity modulation that supports bidirectional mapping between 2D observations and 3D geometry via mesh-based Gaussian splatting in video-to-geometry grounding stage. It jointly considers the absolute and relative position of Gaussian components. Comprehensive experimental evaluations demonstrate that CloDS effectively learns cloth dynamics from visual data while maintaining strong generalization capabilities for unseen configurations. Our code is available at https://github.com/whynot-zyl/CloDS. Visualization results are available at https://github.com/whynot-zyl/CloDS_video}.%\footnote{As in this example.
From Macro to Micro: Benchmarking Microscopic Spatial Intelligence on Molecules via Vision-Language Models
Dec 12, 2025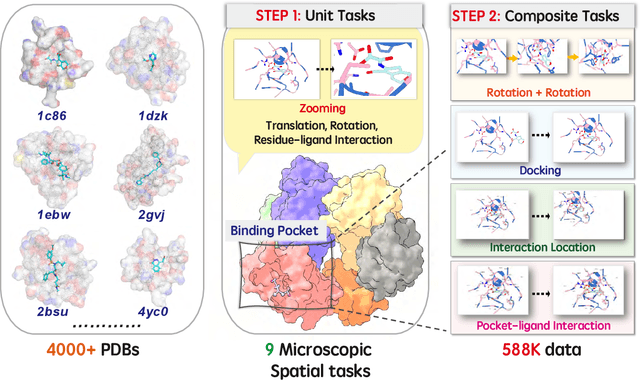
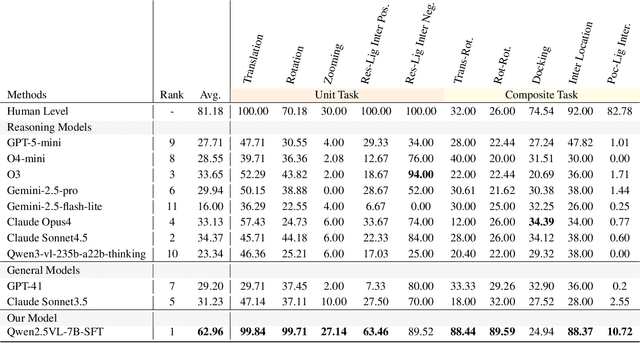
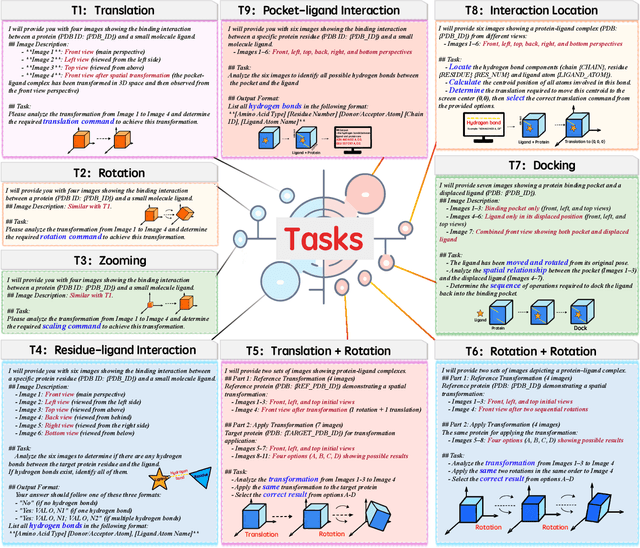
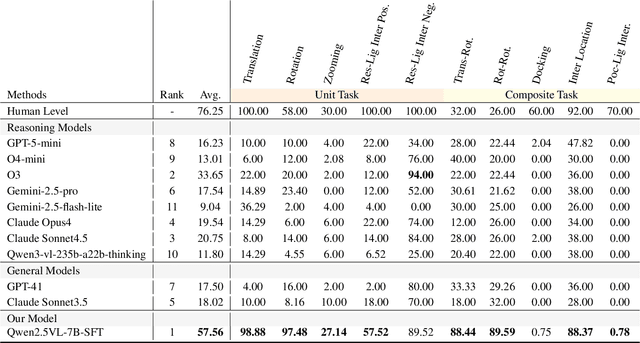
This paper introduces the concept of Microscopic Spatial Intelligence (MiSI), the capability to perceive and reason about the spatial relationships of invisible microscopic entities, which is fundamental to scientific discovery. To assess the potential of Vision-Language Models (VLMs) in this domain, we propose a systematic benchmark framework MiSI-Bench. This framework features over 163,000 question-answer pairs and 587,000 images derived from approximately 4,000 molecular structures, covering nine complementary tasks that evaluate abilities ranging from elementary spatial transformations to complex relational identifications. Experimental results reveal that current state-of-the-art VLMs perform significantly below human level on this benchmark. However, a fine-tuned 7B model demonstrates substantial potential, even surpassing humans in spatial transformation tasks, while its poor performance in scientifically-grounded tasks like hydrogen bond recognition underscores the necessity of integrating explicit domain knowledge for progress toward scientific AGI. The datasets are available at https://huggingface.co/datasets/zongzhao/MiSI-bench.
Equivariant Diffusion for Crystal Structure Prediction
Dec 08, 2025In addressing the challenge of Crystal Structure Prediction (CSP), symmetry-aware deep learning models, particularly diffusion models, have been extensively studied, which treat CSP as a conditional generation task. However, ensuring permutation, rotation, and periodic translation equivariance during diffusion process remains incompletely addressed. In this work, we propose EquiCSP, a novel equivariant diffusion-based generative model. We not only address the overlooked issue of lattice permutation equivariance in existing models, but also develop a unique noising algorithm that rigorously maintains periodic translation equivariance throughout both training and inference processes. Our experiments indicate that EquiCSP significantly surpasses existing models in terms of generating accurate structures and demonstrates faster convergence during the training process.
Fast and Distributed Equivariant Graph Neural Networks by Virtual Node Learning
Jun 24, 2025Equivariant Graph Neural Networks (GNNs) have achieved remarkable success across diverse scientific applications. However, existing approaches face critical efficiency challenges when scaling to large geometric graphs and suffer significant performance degradation when the input graphs are sparsified for computational tractability. To address these limitations, we introduce FastEGNN and DistEGNN, two novel enhancements to equivariant GNNs for large-scale geometric graphs. FastEGNN employs a key innovation: a small ordered set of virtual nodes that effectively approximates the large unordered graph of real nodes. Specifically, we implement distinct message passing and aggregation mechanisms for different virtual nodes to ensure mutual distinctiveness, and minimize Maximum Mean Discrepancy (MMD) between virtual and real coordinates to achieve global distributedness. This design enables FastEGNN to maintain high accuracy while efficiently processing large-scale sparse graphs. For extremely large-scale geometric graphs, we present DistEGNN, a distributed extension where virtual nodes act as global bridges between subgraphs in different devices, maintaining consistency while dramatically reducing memory and computational overhead. We comprehensively evaluate our models across four challenging domains: N-body systems (100 nodes), protein dynamics (800 nodes), Water-3D (8,000 nodes), and our new Fluid113K benchmark (113,000 nodes). Results demonstrate superior efficiency and performance, establishing new capabilities in large-scale equivariant graph learning. Code is available at https://github.com/GLAD-RUC/DistEGNN.
ReasonMed: A 370K Multi-Agent Generated Dataset for Advancing Medical Reasoning
Jun 11, 2025Though reasoning-based large language models (LLMs) have excelled in mathematics and programming, their capabilities in knowledge-intensive medical question answering remain underexplored. To address this, we introduce ReasonMed, the largest medical reasoning dataset, comprising 370k high-quality examples distilled from 1.7 million initial reasoning paths generated by various LLMs. ReasonMed is constructed through a \textit{multi-agent verification and refinement process}, where we design an \textit{Error Refiner} to enhance the reasoning paths by identifying and correcting error-prone steps flagged by a verifier. Leveraging ReasonMed, we systematically investigate best practices for training medical reasoning models and find that combining detailed Chain-of-Thought (CoT) reasoning with concise answer summaries yields the most effective fine-tuning strategy. Based on this strategy, we train ReasonMed-7B, which sets a new benchmark for sub-10B models, outperforming the prior best by 4.17\% and even exceeding LLaMA3.1-70B on PubMedQA by 4.60\%.
STAR-R1: Spatial TrAnsformation Reasoning by Reinforcing Multimodal LLMs
May 26, 2025Multimodal Large Language Models (MLLMs) have demonstrated remarkable capabilities across diverse tasks, yet they lag significantly behind humans in spatial reasoning. We investigate this gap through Transformation-Driven Visual Reasoning (TVR), a challenging task requiring identification of object transformations across images under varying viewpoints. While traditional Supervised Fine-Tuning (SFT) fails to generate coherent reasoning paths in cross-view settings, sparse-reward Reinforcement Learning (RL) suffers from inefficient exploration and slow convergence. To address these limitations, we propose STAR-R1, a novel framework that integrates a single-stage RL paradigm with a fine-grained reward mechanism tailored for TVR. Specifically, STAR-R1 rewards partial correctness while penalizing excessive enumeration and passive inaction, enabling efficient exploration and precise reasoning. Comprehensive evaluations demonstrate that STAR-R1 achieves state-of-the-art performance across all 11 metrics, outperforming SFT by 23% in cross-view scenarios. Further analysis reveals STAR-R1's anthropomorphic behavior and highlights its unique ability to compare all objects for improving spatial reasoning. Our work provides critical insights in advancing the research of MLLMs and reasoning models. The codes, model weights, and data will be publicly available at https://github.com/zongzhao23/STAR-R1.
STAR-R1: Spacial TrAnsformation Reasoning by Reinforcing Multimodal LLMs
May 21, 2025Multimodal Large Language Models (MLLMs) have demonstrated remarkable capabilities across diverse tasks, yet they lag significantly behind humans in spatial reasoning. We investigate this gap through Transformation-Driven Visual Reasoning (TVR), a challenging task requiring identification of object transformations across images under varying viewpoints. While traditional Supervised Fine-Tuning (SFT) fails to generate coherent reasoning paths in cross-view settings, sparse-reward Reinforcement Learning (RL) suffers from inefficient exploration and slow convergence. To address these limitations, we propose STAR-R1, a novel framework that integrates a single-stage RL paradigm with a fine-grained reward mechanism tailored for TVR. Specifically, STAR-R1 rewards partial correctness while penalizing excessive enumeration and passive inaction, enabling efficient exploration and precise reasoning. Comprehensive evaluations demonstrate that STAR-R1 achieves state-of-the-art performance across all 11 metrics, outperforming SFT by 23% in cross-view scenarios. Further analysis reveals STAR-R1's anthropomorphic behavior and highlights its unique ability to compare all objects for improving spatial reasoning. Our work provides critical insights in advancing the research of MLLMs and reasoning models. The codes, model weights, and data will be publicly available at https://github.com/zongzhao23/STAR-R1.
A Concise Survey on Lane Topology Reasoning for HD Mapping
Mar 31, 2025



Lane topology reasoning techniques play a crucial role in high-definition (HD) mapping and autonomous driving applications. While recent years have witnessed significant advances in this field, there has been limited effort to consolidate these works into a comprehensive overview. This survey systematically reviews the evolution and current state of lane topology reasoning methods, categorizing them into three major paradigms: procedural modeling-based methods, aerial imagery-based methods, and onboard sensors-based methods. We analyze the progression from early rule-based approaches to modern learning-based solutions utilizing transformers, graph neural networks (GNNs), and other deep learning architectures. The paper examines standardized evaluation metrics, including road-level measures (APLS and TLTS score), and lane-level metrics (DET and TOP score), along with performance comparisons on benchmark datasets such as OpenLane-V2. We identify key technical challenges, including dataset availability and model efficiency, and outline promising directions for future research. This comprehensive review provides researchers and practitioners with insights into the theoretical frameworks, practical implementations, and emerging trends in lane topology reasoning for HD mapping applications.