 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtein Autoregressive Modeling via Multiscale Structure Generation
Feb 04, 2026We present protein autoregressive modeling (PAR), the first multi-scale autoregressive framework for protein backbone generation via coarse-to-fine next-scale prediction. Using the hierarchical nature of proteins, PAR generates structures that mimic sculpting a statue, forming a coarse topology and refining structural details over scales. To achieve this, PAR consists of three key components: (i) multi-scale downsampling operations that represent protein structures across multiple scales during training; (ii) an autoregressive transformer that encodes multi-scale information and produces conditional embeddings to guide structure generation; (iii) a flow-based backbone decoder that generates backbone atoms conditioned on these embeddings. Moreover, autoregressive models suffer from exposure bias, caused by the training and the generation procedure mismatch, and substantially degrades structure generation quality. We effectively alleviate this issue by adopting noisy context learning and scheduled sampling, enabling robust backbone generation. Notably, PAR exhibits strong zero-shot generalization, supporting flexible human-prompted conditional generation and motif scaffolding without requiring fine-tuning. On the unconditional generation benchmark, PAR effectively learns protein distributions and produces backbones of high design quality, and exhibits favorable scaling behavior. Together, these properties establish PAR as a promising framework for protein structure generation.
Designing Cyclic Peptides via Harmonic SDE with Atom-Bond Modeling
May 27, 2025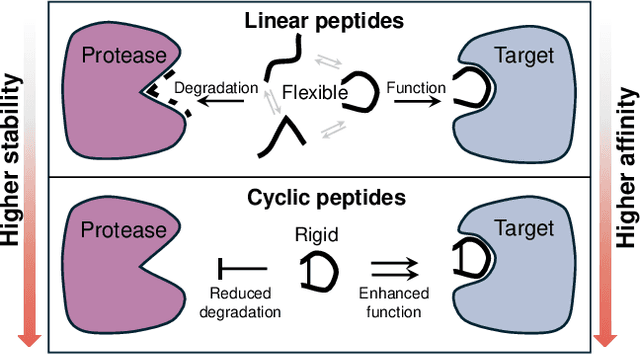
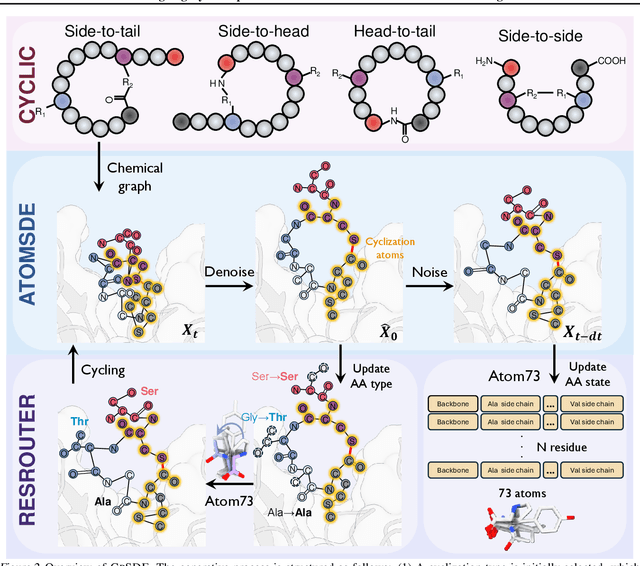
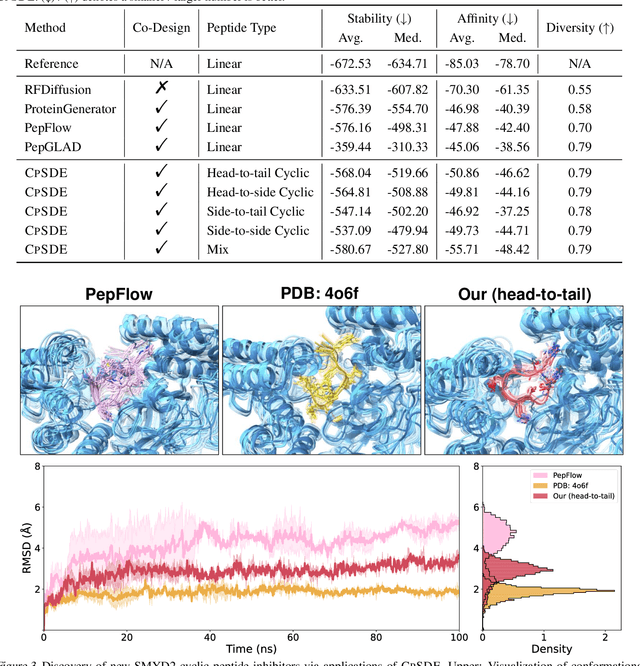
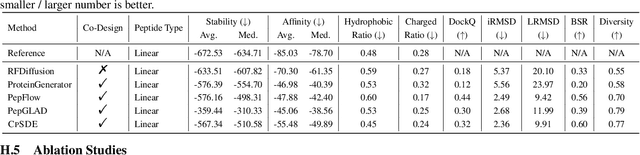
Cyclic peptides offer inherent advantages in pharmaceuticals. For example, cyclic peptides are more resistant to enzymatic hydrolysis compared to linear peptides and usually exhibit excellent stability and affinity. Although deep generative models have achieved great success in linear peptide design, several challenges prevent the development of computational methods for designing diverse types of cyclic peptides. These challenges include the scarcity of 3D structural data on target proteins and associated cyclic peptide ligands, the geometric constraints that cyclization imposes, and the involvement of non-canonical amino acids in cyclization. To address the above challenges, we introduce CpSDE, which consists of two key components: AtomSDE, a generative structure prediction model based on harmonic SDE, and ResRouter, a residue type predictor. Utilizing a routed sampling algorithm that alternates between these two models to iteratively update sequences and structures, CpSDE facilitates the generation of cyclic peptides. By employing explicit all-atom and bond modeling, CpSDE overcomes existing data limitations and is proficient in designing a wide variety of cyclic peptides. Our experimental results demonstrate that the cyclic peptides designed by our method exhibit reliable stability and affinity.
An All-Atom Generative Model for Designing Protein Complexes
Apr 17, 2025Proteins typically exist in complexes, interacting with other proteins or biomolecules to perform their specific biological roles. Research on single-chain protein modeling has been extensively and deeply explored, with advancements seen in models like the series of ESM and AlphaFold. Despite these developments, the study and modeling of multi-chain proteins remain largely uncharted, though they are vital for understanding biological functions. Recognizing the importance of these interactions, we introduce APM (All-Atom Protein Generative Model), a model specifically designed for modeling multi-chain proteins. By integrating atom-level information and leveraging data on multi-chain proteins, APM is capable of precisely modeling inter-chain interactions and designing protein complexes with binding capabilities from scratch. It also performs folding and inverse-folding tasks for multi-chain proteins. Moreover, APM demonstrates versatility in downstream applications: it achieves enhanced performance through supervised fine-tuning (SFT) while also supporting zero-shot sampling in certain tasks, achieving state-of-the-art results. Code will be released at https://github.com/bytedance/apm.
Elucidating the Design Space of Multimodal Protein Language Models
Apr 16, 2025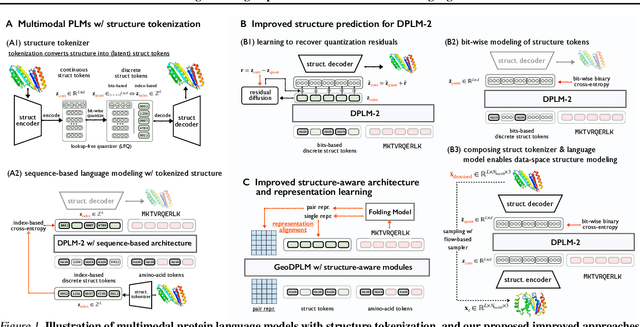
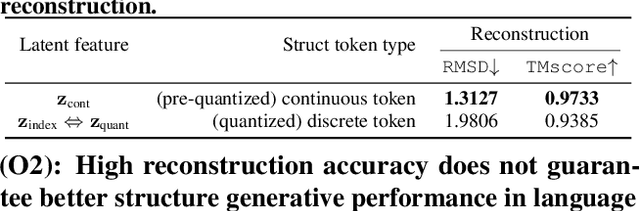
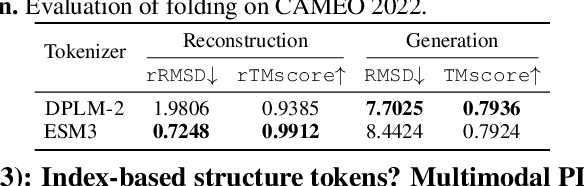
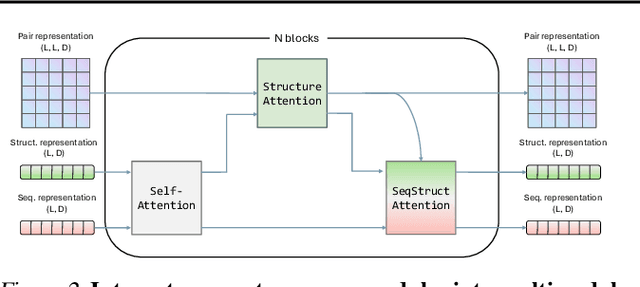
Multimodal protein language models (PLMs) integrate sequence and token-based structural information, serving as a powerful foundation for protein modeling, generation, and design. However, the reliance on tokenizing 3D structures into discrete tokens causes substantial loss of fidelity about fine-grained structural details and correlations. In this paper, we systematically elucidate the design space of multimodal PLMs to overcome their limitations. We identify tokenization loss and inaccurate structure token predictions by the PLMs as major bottlenecks. To address these, our proposed design space covers improved generative modeling, structure-aware architectures and representation learning, and data exploration. Our advancements approach finer-grained supervision, demonstrating that token-based multimodal PLMs can achieve robust structural modeling. The effective design methods dramatically improve the structure generation diversity, and notably, folding abilities of our 650M model by reducing the RMSD from 5.52 to 2.36 on PDB testset, even outperforming 3B baselines and on par with the specialized folding models.
Multilingual Non-Autoregressive Machine Translation without Knowledge Distillation
Feb 06, 2025



Multilingual neural machine translation (MNMT) aims at using one single model for multiple translation directions. Recent work applies non-autoregressive Transformers to improve the efficiency of MNMT, but requires expensive knowledge distillation (KD) processes. To this end, we propose an M-DAT approach to non-autoregressive multilingual machine translation. Our system leverages the recent advance of the directed acyclic Transformer (DAT), which does not require KD. We further propose a pivot back-translation (PivotBT) approach to improve the generalization to unseen translation directions. Experiments show that our M-DAT achieves state-of-the-art performance in non-autoregressive MNMT.
ProteinWeaver: A Divide-and-Assembly Approach for Protein Backbone Design
Nov 08, 2024Nature creates diverse proteins through a `divide and assembly' strategy. Inspired by this idea, we introduce ProteinWeaver, a two-stage framework for protein backbone design. Our method first generates individual protein domains and then employs an SE(3) diffusion model to flexibly assemble these domains. A key challenge lies in the assembling step, given the complex and rugged nature of the inter-domain interaction landscape. To address this challenge, we employ preference alignment to discern complex relationships between structure and interaction landscapes through comparative analysis of generated samples. Comprehensive experiments demonstrate that ProteinWeaver: (1) generates high-quality, novel protein backbones through versatile domain assembly; (2) outperforms RFdiffusion, the current state-of-the-art in backbone design, by 13\% and 39\% for long-chain proteins; (3) shows the potential for cooperative function design through illustrative case studies. To sum up, by introducing a `divide-and-assembly' paradigm, ProteinWeaver advances protein engineering and opens new avenues for functional protein design.
DPLM-2: A Multimodal Diffusion Protein Language Model
Oct 17, 2024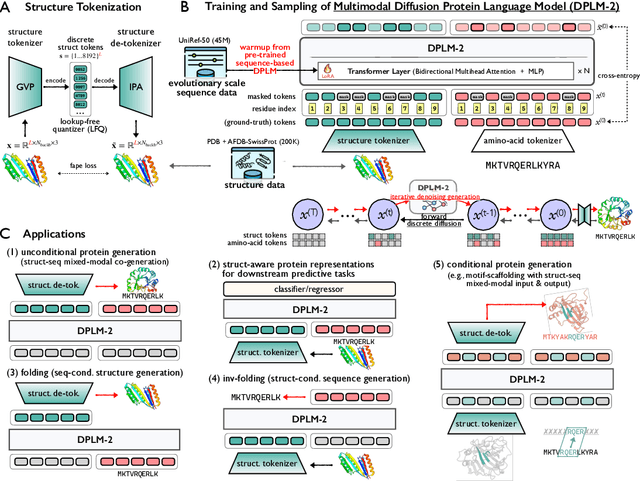
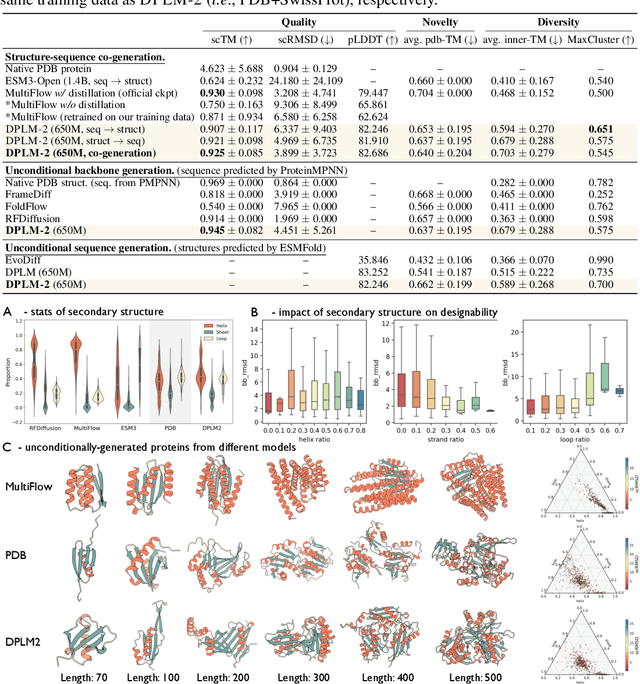
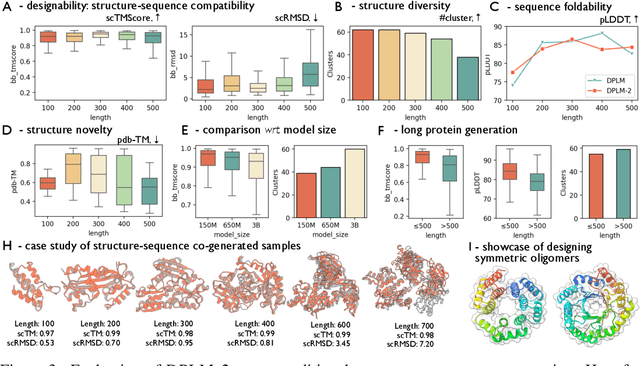
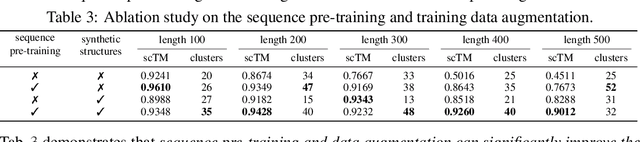
Proteins are essential macromolecules defined by their amino acid sequences, which determine their three-dimensional structures and, consequently, their functions in all living organisms. Therefore, generative protein modeling necessitates a multimodal approach to simultaneously model, understand, and generate both sequences and structures. However, existing methods typically use separate models for each modality, limiting their ability to capture the intricate relationships between sequence and structure. This results in suboptimal performance in tasks that requires joint understanding and generation of both modalities. In this paper, we introduce DPLM-2, a multimodal protein foundation model that extends discrete diffusion protein language model (DPLM) to accommodate both sequences and structures. To enable structural learning with the language model, 3D coordinates are converted to discrete tokens using a lookup-free quantization-based tokenizer. By training on both experimental and high-quality synthetic structures, DPLM-2 learns the joint distribution of sequence and structure, as well as their marginals and conditionals. We also implement an efficient warm-up strategy to exploit the connection between large-scale evolutionary data and structural inductive biases from pre-trained sequence-based protein language models. Empirical evaluation shows that DPLM-2 can simultaneously generate highly compatible amino acid sequences and their corresponding 3D structures eliminating the need for a two-stage generation approach. Moreover, DPLM-2 demonstrates competitive performance in various conditional generation tasks, including folding, inverse folding, and scaffolding with multimodal motif inputs, as well as providing structure-aware representations for predictive tasks.
ProteinBench: A Holistic Evaluation of Protein Foundation Models
Sep 10, 2024Recent years have witnessed a surge in the development of protein foundation models, significantly improving performance in protein prediction and generative tasks ranging from 3D structure prediction and protein design to conformational dynamics. However, the capabilities and limitations associated with these models remain poorly understood due to the absence of a unified evaluation framework. To fill this gap, we introduce ProteinBench, a holistic evaluation framework designed to enhance the transparency of protein foundation models. Our approach consists of three key components: (i) A taxonomic classification of tasks that broadly encompass the main challenges in the protein domain, based on the relationships between different protein modalities; (ii) A multi-metric evaluation approach that assesses performance across four key dimensions: quality, novelty, diversity, and robustness; and (iii) In-depth analyses from various user objectives, providing a holistic view of model performance. Our comprehensive evaluation of protein foundation models reveals several key findings that shed light on their current capabilities and limitations. To promote transparency and facilitate further research, we release the evaluation dataset, code, and a public leaderboard publicly for further analysis and a general modular toolkit. We intend for ProteinBench to be a living benchmark for establishing a standardized, in-depth evaluation framework for protein foundation models, driving their development and application while fostering collaboration within the field.
Antigen-Specific Antibody Design via Direct Energy-based Preference Optimization
Mar 25, 2024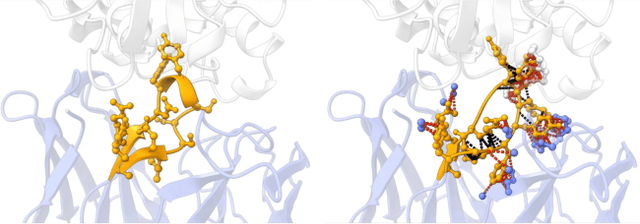
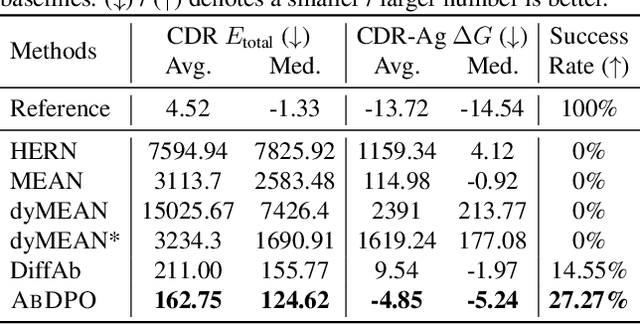
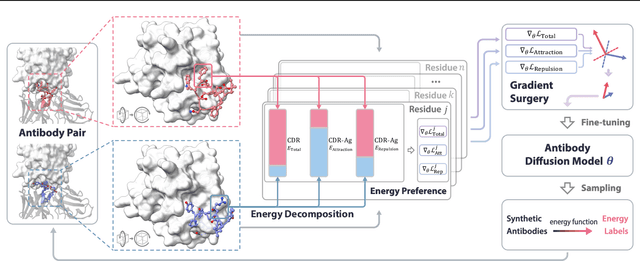
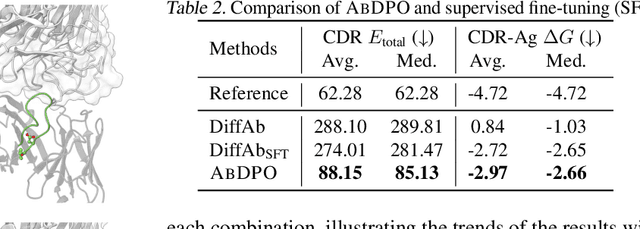
Antibody design, a crucial task with significant implications across various disciplines such as therapeutics and biology, presents considerable challenges due to its intricate nature. In this paper, we tackle antigen-specific antibody design as a protein sequence-structure co-design problem, considering both rationality and functionality. Leveraging a pre-trained conditional diffusion model that jointly models sequences and structures of complementarity-determining regions (CDR) in antibodies with equivariant neural networks, we propose direct energy-based preference optimization to guide the generation of antibodies with both rational structures and considerable binding affinities to given antigens. Our method involves fine-tuning the pre-trained diffusion model using a residue-level decomposed energy preference. Additionally, we employ gradient surgery to address conflicts between various types of energy, such as attraction and repulsion. Experiments on RAbD benchmark show that our approach effectively optimizes the energy of generated antibodies and achieves state-of-the-art performance in designing high-quality antibodies with low total energy and high binding affinity, demonstrating the superiority of our approach.
Diffusion Language Models Are Versatile Protein Learners
Feb 28, 2024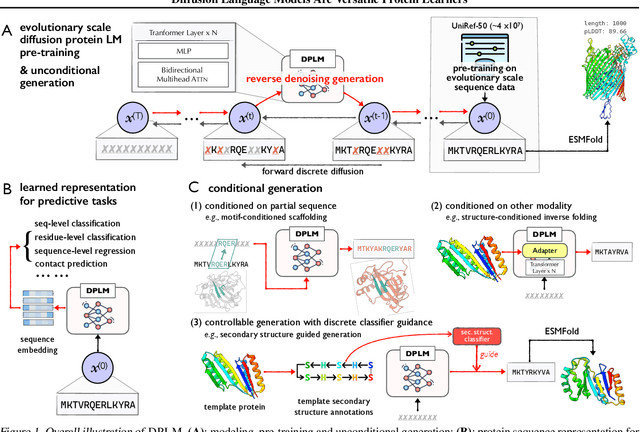
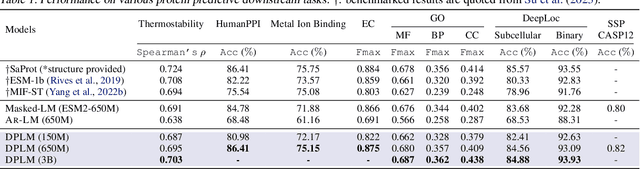
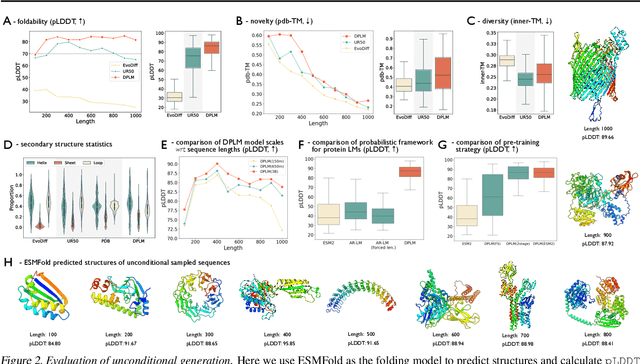
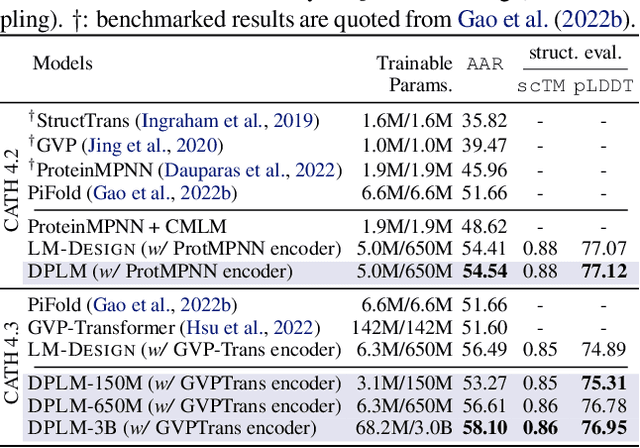
This paper introduces diffusion protein language model (DPLM), a versatile protein language model that demonstrates strong generative and predictive capabilities for protein sequences. We first pre-train scalable DPLMs from evolutionary-scale protein sequences within a generative self-supervised discrete diffusion probabilistic framework, which generalizes language modeling for proteins in a principled way. After pre-training, DPLM exhibits the ability to generate structurally plausible, novel, and diverse protein sequences for unconditional generation. We further demonstrate the proposed diffusion generative pre-training makes DPLM possess a better understanding of proteins, making it a superior representation learner, which can be fine-tuned for various predictive tasks, comparing favorably to ESM2 (Lin et al., 2022). Moreover, DPLM can be tailored for various needs, which showcases its prowess of conditional generation in several ways: (1) conditioning on partial peptide sequences, e.g., generating scaffolds for functional motifs with high success rate; (2) incorporating other modalities as conditioner, e.g., structure-conditioned generation for inverse folding; and (3) steering sequence generation towards desired properties, e.g., satisfying specified secondary structures, through a plug-and-play classifier guidance.