 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRethinking Genomic Modeling Through Optical Character Recognition
Feb 02, 2026Recent genomic foundation models largely adopt large language model architectures that treat DNA as a one-dimensional token sequence. However, exhaustive sequential reading is structurally misaligned with sparse and discontinuous genomic semantics, leading to wasted computation on low-information background and preventing understanding-driven compression for long contexts. Here, we present OpticalDNA, a vision-based framework that reframes genomic modeling as Optical Character Recognition (OCR)-style document understanding. OpticalDNA renders DNA into structured visual layouts and trains an OCR-capable vision--language model with a \emph{visual DNA encoder} and a \emph{document decoder}, where the encoder produces compact, reconstructible visual tokens for high-fidelity compression. Building on this representation, OpticalDNA defines prompt-conditioned objectives over core genomic primitives-reading, region grounding, subsequence retrieval, and masked span completion-thereby learning layout-aware DNA representations that retain fine-grained genomic information under a reduced effective token budget. Across diverse genomic benchmarks, OpticalDNA consistently outperforms recent baselines; on sequences up to 450k bases, it achieves the best overall performance with nearly $20\times$ fewer effective tokens, and surpasses models with up to $985\times$ more activated parameters while tuning only 256k \emph{trainable} parameters.
Surface-based Molecular Design with Multi-modal Flow Matching
Jan 08, 2026Therapeutic peptides show promise in targeting previously undruggable binding sites, with recent advancements in deep generative models enabling full-atom peptide co-design for specific protein receptors. However, the critical role of molecular surfaces in protein-protein interactions (PPIs) has been underexplored. To bridge this gap, we propose an omni-design peptides generation paradigm, called SurfFlow, a novel surface-based generative algorithm that enables comprehensive co-design of sequence, structure, and surface for peptides. SurfFlow employs a multi-modality conditional flow matching (CFM) architecture to learn distributions of surface geometries and biochemical properties, enhancing peptide binding accuracy. Evaluated on the comprehensive PepMerge benchmark, SurfFlow consistently outperforms full-atom baselines across all metrics. These results highlight the advantages of considering molecular surfaces in de novo peptide discovery and demonstrate the potential of integrating multiple protein modalities for more effective therapeutic peptide discovery.
DeepDR: an integrated deep-learning model web server for drug repositioning
Nov 12, 2025Background: Identifying new indications for approved drugs is a complex and time-consuming process that requires extensive knowledge of pharmacology, clinical data, and advanced computational methods. Recently, deep learning (DL) methods have shown their capability for the accurate prediction of drug repositioning. However, implementing DL-based modeling requires in-depth domain knowledge and proficient programming skills. Results: In this application, we introduce DeepDR, the first integrated platform that combines a variety of established DL-based models for disease- and target-specific drug repositioning tasks. DeepDR leverages invaluable experience to recommend candidate drugs, which covers more than 15 networks and a comprehensive knowledge graph that includes 5.9 million edges across 107 types of relationships connecting drugs, diseases, proteins/genes, pathways, and expression from six existing databases and a large scientific corpus of 24 million PubMed publications. Additionally, the recommended results include detailed descriptions of the recommended drugs and visualize key patterns with interpretability through a knowledge graph. Conclusion: DeepDR is free and open to all users without the requirement of registration. We believe it can provide an easy-to-use, systematic, highly accurate, and computationally automated platform for both experimental and computational scientists.
ImageDDI: Image-enhanced Molecular Motif Sequence Representation for Drug-Drug Interaction Prediction
Aug 11, 2025To mitigate the potential adverse health effects of simultaneous multi-drug use, including unexpected side effects and interactions, accurately identifying and predicting drug-drug interactions (DDIs) is considered a crucial task in the field of deep learning. Although existing methods have demonstrated promising performance, they suffer from the bottleneck of limited functional motif-based representation learning, as DDIs are fundamentally caused by motif interactions rather than the overall drug structures. In this paper, we propose an Image-enhanced molecular motif sequence representation framework for \textbf{DDI} prediction, called ImageDDI, which represents a pair of drugs from both global and local structures. Specifically, ImageDDI tokenizes molecules into functional motifs. To effectively represent a drug pair, their motifs are combined into a single sequence and embedded using a transformer-based encoder, starting from the local structure representation. By leveraging the associations between drug pairs, ImageDDI further enhances the spatial representation of molecules using global molecular image information (e.g. texture, shadow, color, and planar spatial relationships). To integrate molecular visual information into functional motif sequence, ImageDDI employs Adaptive Feature Fusion, enhancing the generalization of ImageDDI by dynamically adapting the fusion process of feature representations. Experimental results on widely used datasets demonstrate that ImageDDI outperforms state-of-the-art methods. Moreover, extensive experiments show that ImageDDI achieved competitive performance in both 2D and 3D image-enhanced scenarios compared to other models.
AdaptMol: Adaptive Fusion from Sequence String to Topological Structure for Few-shot Drug Discovery
May 17, 2025



Accurate molecular property prediction (MPP) is a critical step in modern drug development. However, the scarcity of experimental validation data poses a significant challenge to AI-driven research paradigms. Under few-shot learning scenarios, the quality of molecular representations directly dictates the theoretical upper limit of model performance. We present AdaptMol, a prototypical network integrating Adaptive multimodal fusion for Molecular representation. This framework employs a dual-level attention mechanism to dynamically integrate global and local molecular features derived from two modalities: SMILES sequences and molecular graphs. (1) At the local level, structural features such as atomic interactions and substructures are extracted from molecular graphs, emphasizing fine-grained topological information; (2) At the global level, the SMILES sequence provides a holistic representation of the molecule. To validate the necessity of multimodal adaptive fusion, we propose an interpretable approach based on identifying molecular active substructures to demonstrate that multimodal adaptive fusion can efficiently represent molecules. Extensive experiments on three commonly used benchmarks under 5-shot and 10-shot settings demonstrate that AdaptMol achieves state-of-the-art performance in most cases. The rationale-extracted method guides the fusion of two modalities and highlights the importance of both modalities.
EDBench: Large-Scale Electron Density Data for Molecular Modeling
May 14, 2025Existing molecular machine learning force fields (MLFFs) generally focus on the learning of atoms, molecules, and simple quantum chemical properties (such as energy and force), but ignore the importance of electron density (ED) $\rho(r)$ in accurately understanding molecular force fields (MFFs). ED describes the probability of finding electrons at specific locations around atoms or molecules, which uniquely determines all ground state properties (such as energy, molecular structure, etc.) of interactive multi-particle systems according to the Hohenberg-Kohn theorem. However, the calculation of ED relies on the time-consuming first-principles density functional theory (DFT) which leads to the lack of large-scale ED data and limits its application in MLFFs. In this paper, we introduce EDBench, a large-scale, high-quality dataset of ED designed to advance learning-based research at the electronic scale. Built upon the PCQM4Mv2, EDBench provides accurate ED data, covering 3.3 million molecules. To comprehensively evaluate the ability of models to understand and utilize electronic information, we design a suite of ED-centric benchmark tasks spanning prediction, retrieval, and generation. Our evaluation on several state-of-the-art methods demonstrates that learning from EDBench is not only feasible but also achieves high accuracy. Moreover, we show that learning-based method can efficiently calculate ED with comparable precision while significantly reducing the computational cost relative to traditional DFT calculations. All data and benchmarks from EDBench will be freely available, laying a robust foundation for ED-driven drug discovery and materials science.
Enhancing Chemical Reaction and Retrosynthesis Prediction with Large Language Model and Dual-task Learning
May 05, 2025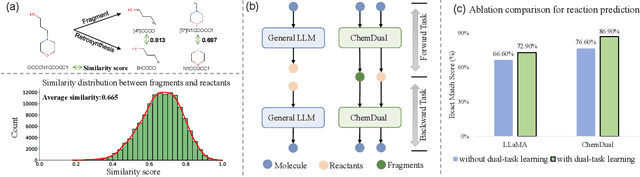
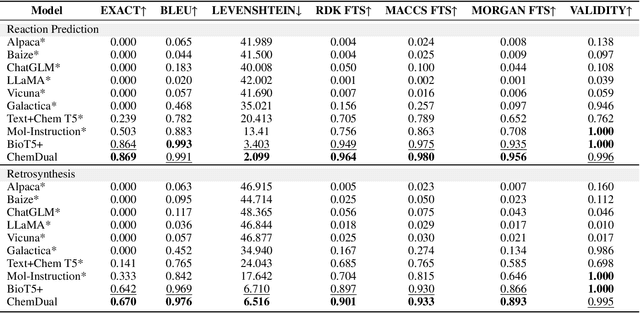


Chemical reaction and retrosynthesis prediction are fundamental tasks in drug discovery. Recently, large language models (LLMs) have shown potential in many domains. However, directly applying LLMs to these tasks faces two major challenges: (i) lacking a large-scale chemical synthesis-related instruction dataset; (ii) ignoring the close correlation between reaction and retrosynthesis prediction for the existing fine-tuning strategies. To address these challenges, we propose ChemDual, a novel LLM framework for accurate chemical synthesis. Specifically, considering the high cost of data acquisition for reaction and retrosynthesis, ChemDual regards the reaction-and-retrosynthesis of molecules as a related recombination-and-fragmentation process and constructs a large-scale of 4.4 million instruction dataset. Furthermore, ChemDual introduces an enhanced LLaMA, equipped with a multi-scale tokenizer and dual-task learning strategy, to jointly optimize the process of recombination and fragmentation as well as the tasks between reaction and retrosynthesis prediction. Extensive experiments on Mol-Instruction and USPTO-50K datasets demonstrate that ChemDual achieves state-of-the-art performance in both predictions of reaction and retrosynthesis, outperforming the existing conventional single-task approaches and the general open-source LLMs. Through molecular docking analysis, ChemDual generates compounds with diverse and strong protein binding affinity, further highlighting its strong potential in drug design.
An All-Atom Generative Model for Designing Protein Complexes
Apr 17, 2025Proteins typically exist in complexes, interacting with other proteins or biomolecules to perform their specific biological roles. Research on single-chain protein modeling has been extensively and deeply explored, with advancements seen in models like the series of ESM and AlphaFold. Despite these developments, the study and modeling of multi-chain proteins remain largely uncharted, though they are vital for understanding biological functions. Recognizing the importance of these interactions, we introduce APM (All-Atom Protein Generative Model), a model specifically designed for modeling multi-chain proteins. By integrating atom-level information and leveraging data on multi-chain proteins, APM is capable of precisely modeling inter-chain interactions and designing protein complexes with binding capabilities from scratch. It also performs folding and inverse-folding tasks for multi-chain proteins. Moreover, APM demonstrates versatility in downstream applications: it achieves enhanced performance through supervised fine-tuning (SFT) while also supporting zero-shot sampling in certain tasks, achieving state-of-the-art results. Code will be released at https://github.com/bytedance/apm.
Property Enhanced Instruction Tuning for Multi-task Molecule Generation with Large Language Models
Dec 24, 2024Large language models (LLMs) are widely applied in various natural language processing tasks such as question answering and machine translation. However, due to the lack of labeled data and the difficulty of manual annotation for biochemical properties, the performance for molecule generation tasks is still limited, especially for tasks involving multi-properties constraints. In this work, we present a two-step framework PEIT (Property Enhanced Instruction Tuning) to improve LLMs for molecular-related tasks. In the first step, we use textual descriptions, SMILES, and biochemical properties as multimodal inputs to pre-train a model called PEIT-GEN, by aligning multi-modal representations to synthesize instruction data. In the second step, we fine-tune existing open-source LLMs with the synthesized data, the resulting PEIT-LLM can handle molecule captioning, text-based molecule generation, molecular property prediction, and our newly proposed multi-constraint molecule generation tasks. Experimental results show that our pre-trained PEIT-GEN outperforms MolT5 and BioT5 in molecule captioning, demonstrating modalities align well between textual descriptions, structures, and biochemical properties. Furthermore, PEIT-LLM shows promising improvements in multi-task molecule generation, proving the scalability of the PEIT framework for various molecular tasks. We release the code, constructed instruction data, and model checkpoints in https://github.com/chenlong164/PEIT.
S$^2$DN: Learning to Denoise Unconvincing Knowledge for Inductive Knowledge Graph Completion
Dec 20, 2024Inductive Knowledge Graph Completion (KGC) aims to infer missing facts between newly emerged entities within knowledge graphs (KGs), posing a significant challenge. While recent studies have shown promising results in inferring such entities through knowledge subgraph reasoning, they suffer from (i) the semantic inconsistencies of similar relations, and (ii) noisy interactions inherent in KGs due to the presence of unconvincing knowledge for emerging entities. To address these challenges, we propose a Semantic Structure-aware Denoising Network (S$^2$DN) for inductive KGC. Our goal is to learn adaptable general semantics and reliable structures to distill consistent semantic knowledge while preserving reliable interactions within KGs. Specifically, we introduce a semantic smoothing module over the enclosing subgraphs to retain the universal semantic knowledge of relations. We incorporate a structure refining module to filter out unreliable interactions and offer additional knowledge, retaining robust structure surrounding target links. Extensive experiments conducted on three benchmark KGs demonstrate that S$^2$DN surpasses the performance of state-of-the-art models. These results demonstrate the effectiveness of S$^2$DN in preserving semantic consistency and enhancing the robustness of filtering out unreliable interactions in contaminated KGs.