 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImageDDI: Image-enhanced Molecular Motif Sequence Representation for Drug-Drug Interaction Prediction
Aug 11, 2025To mitigate the potential adverse health effects of simultaneous multi-drug use, including unexpected side effects and interactions, accurately identifying and predicting drug-drug interactions (DDIs) is considered a crucial task in the field of deep learning. Although existing methods have demonstrated promising performance, they suffer from the bottleneck of limited functional motif-based representation learning, as DDIs are fundamentally caused by motif interactions rather than the overall drug structures. In this paper, we propose an Image-enhanced molecular motif sequence representation framework for \textbf{DDI} prediction, called ImageDDI, which represents a pair of drugs from both global and local structures. Specifically, ImageDDI tokenizes molecules into functional motifs. To effectively represent a drug pair, their motifs are combined into a single sequence and embedded using a transformer-based encoder, starting from the local structure representation. By leveraging the associations between drug pairs, ImageDDI further enhances the spatial representation of molecules using global molecular image information (e.g. texture, shadow, color, and planar spatial relationships). To integrate molecular visual information into functional motif sequence, ImageDDI employs Adaptive Feature Fusion, enhancing the generalization of ImageDDI by dynamically adapting the fusion process of feature representations. Experimental results on widely used datasets demonstrate that ImageDDI outperforms state-of-the-art methods. Moreover, extensive experiments show that ImageDDI achieved competitive performance in both 2D and 3D image-enhanced scenarios compared to other models.
S$^2$DN: Learning to Denoise Unconvincing Knowledge for Inductive Knowledge Graph Completion
Dec 20, 2024Inductive Knowledge Graph Completion (KGC) aims to infer missing facts between newly emerged entities within knowledge graphs (KGs), posing a significant challenge. While recent studies have shown promising results in inferring such entities through knowledge subgraph reasoning, they suffer from (i) the semantic inconsistencies of similar relations, and (ii) noisy interactions inherent in KGs due to the presence of unconvincing knowledge for emerging entities. To address these challenges, we propose a Semantic Structure-aware Denoising Network (S$^2$DN) for inductive KGC. Our goal is to learn adaptable general semantics and reliable structures to distill consistent semantic knowledge while preserving reliable interactions within KGs. Specifically, we introduce a semantic smoothing module over the enclosing subgraphs to retain the universal semantic knowledge of relations. We incorporate a structure refining module to filter out unreliable interactions and offer additional knowledge, retaining robust structure surrounding target links. Extensive experiments conducted on three benchmark KGs demonstrate that S$^2$DN surpasses the performance of state-of-the-art models. These results demonstrate the effectiveness of S$^2$DN in preserving semantic consistency and enhancing the robustness of filtering out unreliable interactions in contaminated KGs.
KGExplainer: Towards Exploring Connected Subgraph Explanations for Knowledge Graph Completion
Apr 05, 2024


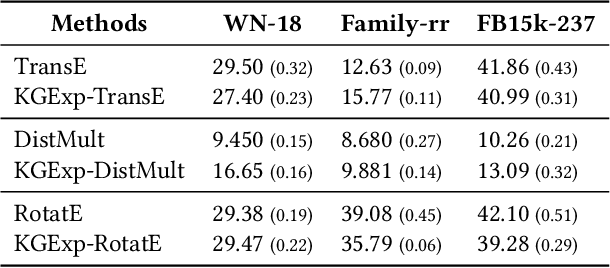
Knowledge graph completion (KGC) aims to alleviate the inherent incompleteness of knowledge graphs (KGs), which is a critical task for various applications, such as recommendations on the web. Although knowledge graph embedding (KGE) models have demonstrated superior predictive performance on KGC tasks, these models infer missing links in a black-box manner that lacks transparency and accountability, preventing researchers from developing accountable models. Existing KGE-based explanation methods focus on exploring key paths or isolated edges as explanations, which is information-less to reason target prediction. Additionally, the missing ground truth leads to these explanation methods being ineffective in quantitatively evaluating explored explanations. To overcome these limitations, we propose KGExplainer, a model-agnostic method that identifies connected subgraph explanations and distills an evaluator to assess them quantitatively. KGExplainer employs a perturbation-based greedy search algorithm to find key connected subgraphs as explanations within the local structure of target predictions. To evaluate the quality of the explored explanations, KGExplainer distills an evaluator from the target KGE model. By forwarding the explanations to the evaluator, our method can examine the fidelity of them. Extensive experiments on benchmark datasets demonstrate that KGExplainer yields promising improvement and achieves an optimal ratio of 83.3% in human evaluation.
Learning to Denoise Unreliable Interactions for Link Prediction on Biomedical Knowledge Graph
Dec 09, 2023



Link prediction in biomedical knowledge graphs (KGs) aims at predicting unknown interactions between entities, including drug-target interaction (DTI) and drug-drug interaction (DDI), which is critical for drug discovery and therapeutics. Previous methods prefer to utilize the rich semantic relations and topological structure of the KG to predict missing links, yielding promising outcomes. However, all these works only focus on improving the predictive performance without considering the inevitable noise and unreliable interactions existing in the KGs, which limits the development of KG-based computational methods. To address these limitations, we propose a Denoised Link Prediction framework, called DenoisedLP. DenoisedLP obtains reliable interactions based on the local subgraph by denoising noisy links in a learnable way, providing a universal module for mining underlying task-relevant relations. To collaborate with the smoothed semantic information, DenoisedLP introduces the semantic subgraph by blurring conflict relations around the predicted link. By maximizing the mutual information between the reliable structure and smoothed semantic relations, DenoisedLP emphasizes the informative interactions for predicting relation-specific links. Experimental results on real-world datasets demonstrate that DenoisedLP outperforms state-of-the-art methods on DTI and DDI prediction tasks, and verify the effectiveness and robustness of denoising unreliable interactions on the contaminated KGs.
Comprehensive evaluation of deep and graph learning on drug-drug interactions prediction
Jun 08, 2023Recent advances and achievements of artificial intelligence (AI) as well as deep and graph learning models have established their usefulness in biomedical applications, especially in drug-drug interactions (DDIs). DDIs refer to a change in the effect of one drug to the presence of another drug in the human body, which plays an essential role in drug discovery and clinical research. DDIs prediction through traditional clinical trials and experiments is an expensive and time-consuming process. To correctly apply the advanced AI and deep learning, the developer and user meet various challenges such as the availability and encoding of data resources, and the design of computational methods. This review summarizes chemical structure based, network based, NLP based and hybrid methods, providing an updated and accessible guide to the broad researchers and development community with different domain knowledge. We introduce widely-used molecular representation and describe the theoretical frameworks of graph neural network models for representing molecular structures. We present the advantages and disadvantages of deep and graph learning methods by performing comparative experiments. We discuss the potential technical challenges and highlight future directions of deep and graph learning models for accelerating DDIs prediction.