 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeERNIE 5.0 Technical Report
Feb 04, 2026In this report, we introduce ERNIE 5.0, a natively autoregressive foundation model desinged for unified multimodal understanding and generation across text, image, video, and audio. All modalities are trained from scratch under a unified next-group-of-tokens prediction objective, based on an ultra-sparse mixture-of-experts (MoE) architecture with modality-agnostic expert routing. To address practical challenges in large-scale deployment under diverse resource constraints, ERNIE 5.0 adopts a novel elastic training paradigm. Within a single pre-training run, the model learns a family of sub-models with varying depths, expert capacities, and routing sparsity, enabling flexible trade-offs among performance, model size, and inference latency in memory- or time-constrained scenarios. Moreover, we systematically address the challenges of scaling reinforcement learning to unified foundation models, thereby guaranteeing efficient and stable post-training under ultra-sparse MoE architectures and diverse multimodal settings. Extensive experiments demonstrate that ERNIE 5.0 achieves strong and balanced performance across multiple modalities. To the best of our knowledge, among publicly disclosed models, ERNIE 5.0 represents the first production-scale realization of a trillion-parameter unified autoregressive model that supports both multimodal understanding and generation. To facilitate further research, we present detailed visualizations of modality-agnostic expert routing in the unified model, alongside comprehensive empirical analysis of elastic training, aiming to offer profound insights to the community.
Patient-Conditioned Adaptive Offsets for Reliable Diagnosis across Subgroups
Jan 19, 2026AI models for medical diagnosis often exhibit uneven performance across patient populations due to heterogeneity in disease prevalence, imaging appearance, and clinical risk profiles. Existing algorithmic fairness approaches typically seek to reduce such disparities by suppressing sensitive attributes. However, in medical settings these attributes often carry essential diagnostic information, and removing them can degrade accuracy and reliability, particularly in high-stakes applications. In contrast, clinical decision making explicitly incorporates patient context when interpreting diagnostic evidence, suggesting a different design direction for subgroup-aware models. In this paper, we introduce HyperAdapt, a patient-conditioned adaptation framework that improves subgroup reliability while maintaining a shared diagnostic model. Clinically relevant attributes such as age and sex are encoded into a compact embedding and used to condition a hypernetwork-style module, which generates small residual modulation parameters for selected layers of a shared backbone. This design preserves the general medical knowledge learned by the backbone while enabling targeted adjustments that reflect patient-specific variability. To ensure efficiency and robustness, adaptations are constrained through low-rank and bottlenecked parameterizations, limiting both model complexity and computational overhead. Experiments across multiple public medical imaging benchmarks demonstrate that the proposed approach consistently improves subgroup-level performance without sacrificing overall accuracy. On the PAD-UFES-20 dataset, our method outperforms the strongest competing baseline by 4.1% in recall and 4.4% in F1 score, with larger gains observed for underrepresented patient populations.
MiMo-V2-Flash Technical Report
Jan 08, 2026We present MiMo-V2-Flash, a Mixture-of-Experts (MoE) model with 309B total parameters and 15B active parameters, designed for fast, strong reasoning and agentic capabilities. MiMo-V2-Flash adopts a hybrid attention architecture that interleaves Sliding Window Attention (SWA) with global attention, with a 128-token sliding window under a 5:1 hybrid ratio. The model is pre-trained on 27 trillion tokens with Multi-Token Prediction (MTP), employing a native 32k context length and subsequently extended to 256k. To efficiently scale post-training compute, MiMo-V2-Flash introduces a novel Multi-Teacher On-Policy Distillation (MOPD) paradigm. In this framework, domain-specialized teachers (e.g., trained via large-scale reinforcement learning) provide dense and token-level reward, enabling the student model to perfectly master teacher expertise. MiMo-V2-Flash rivals top-tier open-weight models such as DeepSeek-V3.2 and Kimi-K2, despite using only 1/2 and 1/3 of their total parameters, respectively. During inference, by repurposing MTP as a draft model for speculative decoding, MiMo-V2-Flash achieves up to 3.6 acceptance length and 2.6x decoding speedup with three MTP layers. We open-source both the model weights and the three-layer MTP weights to foster open research and community collaboration.
Departures: Distributional Transport for Single-Cell Perturbation Prediction with Neural Schrödinger Bridges
Nov 17, 2025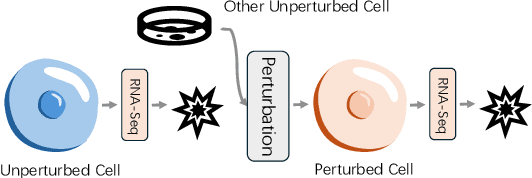
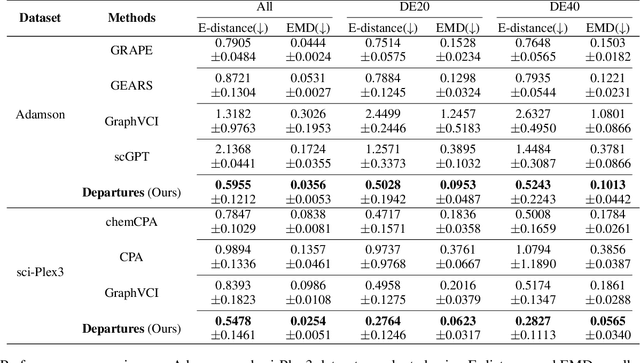
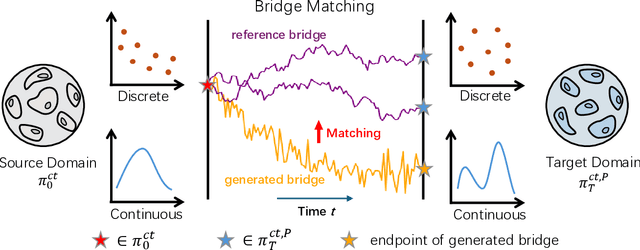

Predicting single-cell perturbation outcomes directly advances gene function analysis and facilitates drug candidate selection, making it a key driver of both basic and translational biomedical research. However, a major bottleneck in this task is the unpaired nature of single-cell data, as the same cell cannot be observed both before and after perturbation due to the destructive nature of sequencing. Although some neural generative transport models attempt to tackle unpaired single-cell perturbation data, they either lack explicit conditioning or depend on prior spaces for indirect distribution alignment, limiting precise perturbation modeling. In this work, we approximate Schrödinger Bridge (SB), which defines stochastic dynamic mappings recovering the entropy-regularized optimal transport (OT), to directly align the distributions of control and perturbed single-cell populations across different perturbation conditions. Unlike prior SB approximations that rely on bidirectional modeling to infer optimal source-target sample coupling, we leverage Minibatch-OT based pairing to avoid such bidirectional inference and the associated ill-posedness of defining the reverse process. This pairing directly guides bridge learning, yielding a scalable approximation to the SB. We approximate two SB models, one modeling discrete gene activation states and the other continuous expression distributions. Joint training enables accurate perturbation modeling and captures single-cell heterogeneity. Experiments on public genetic and drug perturbation datasets show that our model effectively captures heterogeneous single-cell responses and achieves state-of-the-art performance.
PRESCRIBE: Predicting Single-Cell Responses with Bayesian Estimation
Oct 09, 2025In single-cell perturbation prediction, a central task is to forecast the effects of perturbing a gene unseen in the training data. The efficacy of such predictions depends on two factors: (1) the similarity of the target gene to those covered in the training data, which informs model (epistemic) uncertainty, and (2) the quality of the corresponding training data, which reflects data (aleatoric) uncertainty. Both factors are critical for determining the reliability of a prediction, particularly as gene perturbation is an inherently stochastic biochemical process. In this paper, we propose PRESCRIBE (PREdicting Single-Cell Response wIth Bayesian Estimation), a multivariate deep evidential regression framework designed to measure both sources of uncertainty jointly. Our analysis demonstrates that PRESCRIBE effectively estimates a confidence score for each prediction, which strongly correlates with its empirical accuracy. This capability enables the filtering of untrustworthy results, and in our experiments, it achieves steady accuracy improvements of over 3% compared to comparable baselines.
AT-CXR: Uncertainty-Aware Agentic Triage for Chest X-rays
Aug 26, 2025Agentic AI is advancing rapidly, yet truly autonomous medical-imaging triage, where a system decides when to stop, escalate, or defer under real constraints, remains relatively underexplored. To address this gap, we introduce AT-CXR, an uncertainty-aware agent for chest X-rays. The system estimates per-case confidence and distributional fit, then follows a stepwise policy to issue an automated decision or abstain with a suggested label for human intervention. We evaluate two router designs that share the same inputs and actions: a deterministic rule-based router and an LLM-decided router. Across five-fold evaluation on a balanced subset of NIH ChestX-ray14 dataset, both variants outperform strong zero-shot vision-language models and state-of-the-art supervised classifiers, achieving higher full-coverage accuracy and superior selective-prediction performance, evidenced by a lower area under the risk-coverage curve (AURC) and a lower error rate at high coverage, while operating with lower latency that meets practical clinical constraints. The two routers provide complementary operating points, enabling deployments to prioritize maximal throughput or maximal accuracy. Our code is available at https://github.com/XLIAaron/uncertainty-aware-cxr-agent.
Hunyuan-TurboS: Advancing Large Language Models through Mamba-Transformer Synergy and Adaptive Chain-of-Thought
May 21, 2025As Large Language Models (LLMs) rapidly advance, we introduce Hunyuan-TurboS, a novel large hybrid Transformer-Mamba Mixture of Experts (MoE) model. It synergistically combines Mamba's long-sequence processing efficiency with Transformer's superior contextual understanding. Hunyuan-TurboS features an adaptive long-short chain-of-thought (CoT) mechanism, dynamically switching between rapid responses for simple queries and deep "thinking" modes for complex problems, optimizing computational resources. Architecturally, this 56B activated (560B total) parameter model employs 128 layers (Mamba2, Attention, FFN) with an innovative AMF/MF block pattern. Faster Mamba2 ensures linear complexity, Grouped-Query Attention minimizes KV cache, and FFNs use an MoE structure. Pre-trained on 16T high-quality tokens, it supports a 256K context length and is the first industry-deployed large-scale Mamba model. Our comprehensive post-training strategy enhances capabilities via Supervised Fine-Tuning (3M instructions), a novel Adaptive Long-short CoT Fusion method, Multi-round Deliberation Learning for iterative improvement, and a two-stage Large-scale Reinforcement Learning process targeting STEM and general instruction-following. Evaluations show strong performance: overall top 7 rank on LMSYS Chatbot Arena with a score of 1356, outperforming leading models like Gemini-2.0-Flash-001 (1352) and o4-mini-2025-04-16 (1345). TurboS also achieves an average of 77.9% across 23 automated benchmarks. Hunyuan-TurboS balances high performance and efficiency, offering substantial capabilities at lower inference costs than many reasoning models, establishing a new paradigm for efficient large-scale pre-trained models.
EDBench: Large-Scale Electron Density Data for Molecular Modeling
May 14, 2025Existing molecular machine learning force fields (MLFFs) generally focus on the learning of atoms, molecules, and simple quantum chemical properties (such as energy and force), but ignore the importance of electron density (ED) $\rho(r)$ in accurately understanding molecular force fields (MFFs). ED describes the probability of finding electrons at specific locations around atoms or molecules, which uniquely determines all ground state properties (such as energy, molecular structure, etc.) of interactive multi-particle systems according to the Hohenberg-Kohn theorem. However, the calculation of ED relies on the time-consuming first-principles density functional theory (DFT) which leads to the lack of large-scale ED data and limits its application in MLFFs. In this paper, we introduce EDBench, a large-scale, high-quality dataset of ED designed to advance learning-based research at the electronic scale. Built upon the PCQM4Mv2, EDBench provides accurate ED data, covering 3.3 million molecules. To comprehensively evaluate the ability of models to understand and utilize electronic information, we design a suite of ED-centric benchmark tasks spanning prediction, retrieval, and generation. Our evaluation on several state-of-the-art methods demonstrates that learning from EDBench is not only feasible but also achieves high accuracy. Moreover, we show that learning-based method can efficiently calculate ED with comparable precision while significantly reducing the computational cost relative to traditional DFT calculations. All data and benchmarks from EDBench will be freely available, laying a robust foundation for ED-driven drug discovery and materials science.
GRAPE: Heterogeneous Graph Representation Learning for Genetic Perturbation with Coding and Non-Coding Biotype
May 06, 2025Predicting genetic perturbations enables the identification of potentially crucial genes prior to wet-lab experiments, significantly improving overall experimental efficiency. Since genes are the foundation of cellular life, building gene regulatory networks (GRN) is essential to understand and predict the effects of genetic perturbations. However, current methods fail to fully leverage gene-related information, and solely rely on simple evaluation metrics to construct coarse-grained GRN. More importantly, they ignore functional differences between biotypes, limiting the ability to capture potential gene interactions. In this work, we leverage pre-trained large language model and DNA sequence model to extract features from gene descriptions and DNA sequence data, respectively, which serve as the initialization for gene representations. Additionally, we introduce gene biotype information for the first time in genetic perturbation, simulating the distinct roles of genes with different biotypes in regulating cellular processes, while capturing implicit gene relationships through graph structure learning (GSL). We propose GRAPE, a heterogeneous graph neural network (HGNN) that leverages gene representations initialized with features from descriptions and sequences, models the distinct roles of genes with different biotypes, and dynamically refines the GRN through GSL. The results on publicly available datasets show that our method achieves state-of-the-art performance.
Adversarial Curriculum Graph-Free Knowledge Distillation for Graph Neural Networks
Apr 02, 2025Data-free Knowledge Distillation (DFKD) is a method that constructs pseudo-samples using a generator without real data, and transfers knowledge from a teacher model to a student by enforcing the student to overcome dimensional differences and learn to mimic the teacher's outputs on these pseudo-samples. In recent years, various studies in the vision domain have made notable advancements in this area. However, the varying topological structures and non-grid nature of graph data render the methods from the vision domain ineffective. Building upon prior research into differentiable methods for graph neural networks, we propose a fast and high-quality data-free knowledge distillation approach in this paper. Without compromising distillation quality, the proposed graph-free KD method (ACGKD) significantly reduces the spatial complexity of pseudo-graphs by leveraging the Binary Concrete distribution to model the graph structure and introducing a spatial complexity tuning parameter. This approach enables efficient gradient computation for the graph structure, thereby accelerating the overall distillation process. Additionally, ACGKD eliminates the dimensional ambiguity between the student and teacher models by increasing the student's dimensions and reusing the teacher's classifier. Moreover, it equips graph knowledge distillation with a CL-based strategy to ensure the student learns graph structures progressively. Extensive experiments demonstrate that ACGKD achieves state-of-the-art performance in distilling knowledge from GNNs without training data.