 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeNovoBench: Benchmarking Deep Learning-based De Novo Peptide Sequencing Methods in Proteomics
Jun 16, 2024Tandem mass spectrometry has played a pivotal role in advancing proteomics, enabling the high-throughput analysis of protein composition in biological tissues. Many deep learning methods have been developed for \emph{de novo} peptide sequencing task, i.e., predicting the peptide sequence for the observed mass spectrum. However, two key challenges seriously hinder the further advancement of this important task. Firstly, since there is no consensus for the evaluation datasets, the empirical results in different research papers are often not comparable, leading to unfair comparison. Secondly, the current methods are usually limited to amino acid-level or peptide-level precision and recall metrics. In this work, we present the first unified benchmark NovoBench for \emph{de novo} peptide sequencing, which comprises diverse mass spectrum data, integrated models, and comprehensive evaluation metrics. Recent impressive methods, including DeepNovo, PointNovo, Casanovo, InstaNovo, AdaNovo and $\pi$-HelixNovo are integrated into our framework. In addition to amino acid-level and peptide-level precision and recall, we evaluate the models' performance in terms of identifying post-tranlational modifications (PTMs), efficiency and robustness to peptide length, noise peaks and missing fragment ratio, which are important influencing factors while seldom be considered. Leveraging this benchmark, we conduct a large-scale study of current methods, report many insightful findings that open up new possibilities for future development. The benchmark will be open-sourced to facilitate future research and application.
AdaNovo: Adaptive \emph{De Novo} Peptide Sequencing with Conditional Mutual Information
Mar 15, 2024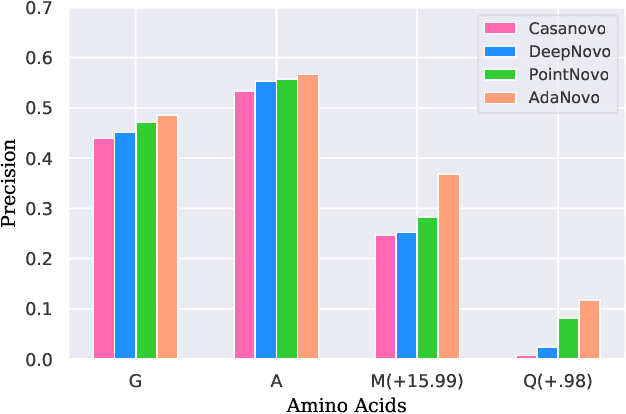
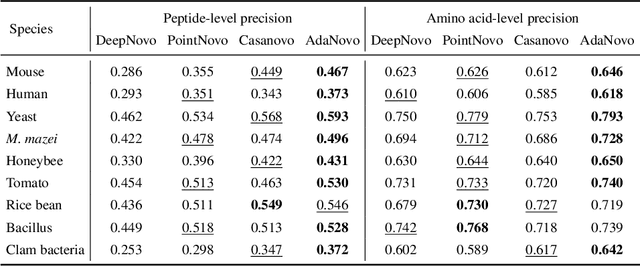
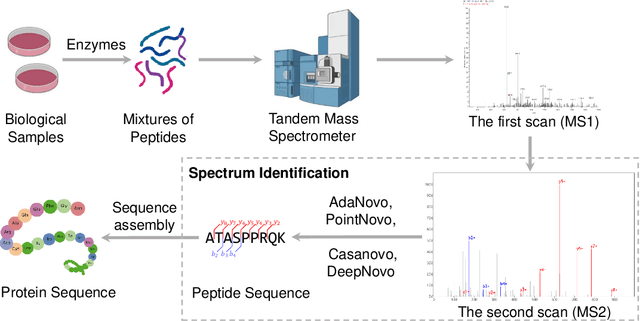
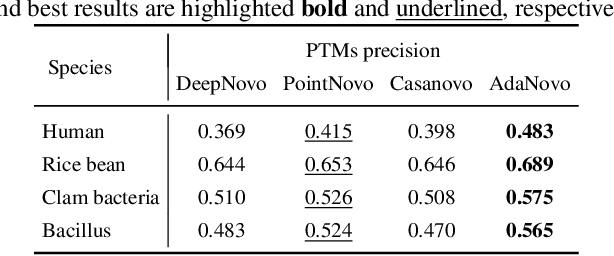
Tandem mass spectrometry has played a pivotal role in advancing proteomics, enabling the analysis of protein composition in biological samples. Despite the development of various deep learning methods for identifying amino acid sequences (peptides) responsible for observed spectra, challenges persist in \emph{de novo} peptide sequencing. Firstly, prior methods struggle to identify amino acids with post-translational modifications (PTMs) due to their lower frequency in training data compared to canonical amino acids, further resulting in decreased peptide-level identification precision. Secondly, diverse types of noise and missing peaks in mass spectra reduce the reliability of training data (peptide-spectrum matches, PSMs). To address these challenges, we propose AdaNovo, a novel framework that calculates conditional mutual information (CMI) between the spectrum and each amino acid/peptide, using CMI for adaptive model training. Extensive experiments demonstrate AdaNovo's state-of-the-art performance on a 9-species benchmark, where the peptides in the training set are almost completely disjoint from the peptides of the test sets. Moreover, AdaNovo excels in identifying amino acids with PTMs and exhibits robustness against data noise. The supplementary materials contain the official code.