 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMeToken: Uniform Micro-environment Token Boosts Post-Translational Modification Prediction
Nov 04, 2024



Post-translational modifications (PTMs) profoundly expand the complexity and functionality of the proteome, regulating protein attributes and interactions that are crucial for biological processes. Accurately predicting PTM sites and their specific types is therefore essential for elucidating protein function and understanding disease mechanisms. Existing computational approaches predominantly focus on protein sequences to predict PTM sites, driven by the recognition of sequence-dependent motifs. However, these approaches often overlook protein structural contexts. In this work, we first compile a large-scale sequence-structure PTM dataset, which serves as the foundation for fair comparison. We introduce the MeToken model, which tokenizes the micro-environment of each amino acid, integrating both sequence and structural information into unified discrete tokens. This model not only captures the typical sequence motifs associated with PTMs but also leverages the spatial arrangements dictated by protein tertiary structures, thus providing a holistic view of the factors influencing PTM sites. Designed to address the long-tail distribution of PTM types, MeToken employs uniform sub-codebooks that ensure even the rarest PTMs are adequately represented and distinguished. We validate the effectiveness and generalizability of MeToken across multiple datasets, demonstrating its superior performance in accurately identifying PTM types. The results underscore the importance of incorporating structural data and highlight MeToken's potential in facilitating accurate and comprehensive PTM predictions, which could significantly impact proteomics research. The code and datasets are available at https://github.com/A4Bio/MeToken.
FlexMol: A Flexible Toolkit for Benchmarking Molecular Relational Learning
Oct 19, 2024



Molecular relational learning (MRL) is crucial for understanding the interaction behaviors between molecular pairs, a critical aspect of drug discovery and development. However, the large feasible model space of MRL poses significant challenges to benchmarking, and existing MRL frameworks face limitations in flexibility and scope. To address these challenges, avoid repetitive coding efforts, and ensure fair comparison of models, we introduce FlexMol, a comprehensive toolkit designed to facilitate the construction and evaluation of diverse model architectures across various datasets and performance metrics. FlexMol offers a robust suite of preset model components, including 16 drug encoders, 13 protein sequence encoders, 9 protein structure encoders, and 7 interaction layers. With its easy-to-use API and flexibility, FlexMol supports the dynamic construction of over 70, 000 distinct combinations of model architectures. Additionally, we provide detailed benchmark results and code examples to demonstrate FlexMol's effectiveness in simplifying and standardizing MRL model development and comparison.
PSC-CPI: Multi-Scale Protein Sequence-Structure Contrasting for Efficient and Generalizable Compound-Protein Interaction Prediction
Feb 13, 2024



Compound-Protein Interaction (CPI) prediction aims to predict the pattern and strength of compound-protein interactions for rational drug discovery. Existing deep learning-based methods utilize only the single modality of protein sequences or structures and lack the co-modeling of the joint distribution of the two modalities, which may lead to significant performance drops in complex real-world scenarios due to various factors, e.g., modality missing and domain shifting. More importantly, these methods only model protein sequences and structures at a single fixed scale, neglecting more fine-grained multi-scale information, such as those embedded in key protein fragments. In this paper, we propose a novel multi-scale Protein Sequence-structure Contrasting framework for CPI prediction (PSC-CPI), which captures the dependencies between protein sequences and structures through both intra-modality and cross-modality contrasting. We further apply length-variable protein augmentation to allow contrasting to be performed at different scales, from the amino acid level to the sequence level. Finally, in order to more fairly evaluate the model generalizability, we split the test data into four settings based on whether compounds and proteins have been observed during the training stage. Extensive experiments have shown that PSC-CPI generalizes well in all four settings, particularly in the more challenging ``Unseen-Both" setting, where neither compounds nor proteins have been observed during training. Furthermore, even when encountering a situation of modality missing, i.e., inference with only single-modality protein data, PSC-CPI still exhibits comparable or even better performance than previous approaches.
A Graph is Worth $K$ Words: Euclideanizing Graph using Pure Transformer
Feb 04, 2024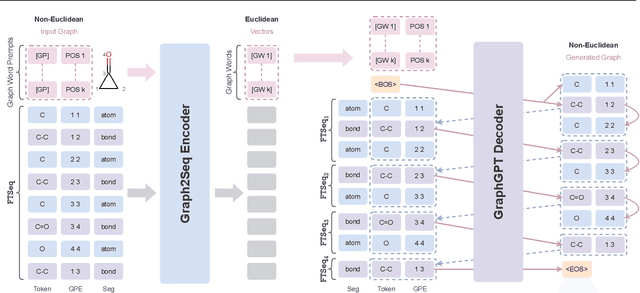
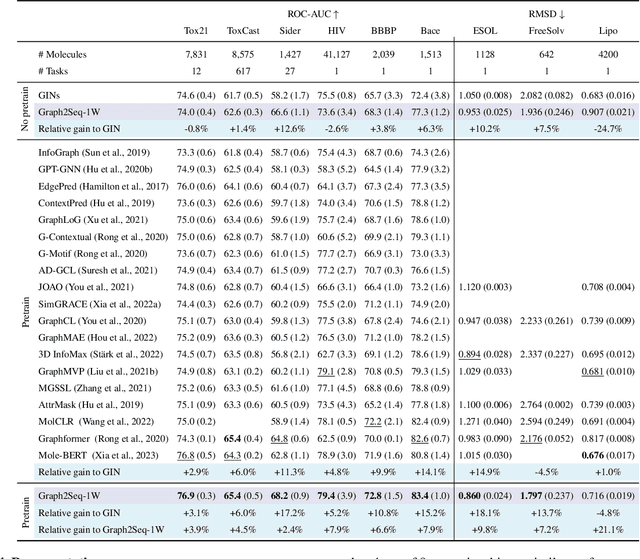
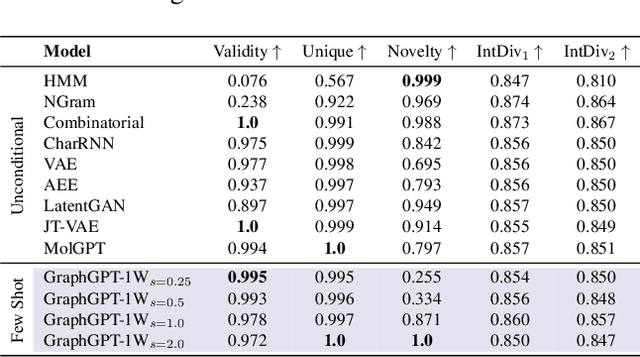

Can we model non-Euclidean graphs as pure language or even Euclidean vectors while retaining their inherent information? The non-Euclidean property have posed a long term challenge in graph modeling. Despite recent GNN and Graphformer efforts encoding graphs as Euclidean vectors, recovering original graph from the vectors remains a challenge. We introduce GraphsGPT, featuring a Graph2Seq encoder that transforms non-Euclidean graphs into learnable graph words in a Euclidean space, along with a GraphGPT decoder that reconstructs the original graph from graph words to ensure information equivalence. We pretrain GraphsGPT on 100M molecules and yield some interesting findings: (1) Pretrained Graph2Seq excels in graph representation learning, achieving state-of-the-art results on 8/9 graph classification and regression tasks. (2) Pretrained GraphGPT serves as a strong graph generator, demonstrated by its ability to perform both unconditional and conditional graph generation. (3) Graph2Seq+GraphGPT enables effective graph mixup in the Euclidean space, overcoming previously known non-Euclidean challenge. (4) Our proposed novel edge-centric GPT pretraining task is effective in graph fields, underscoring its success in both representation and generation.
Deep Manifold Graph Auto-Encoder for Attributed Graph Embedding
Jan 12, 2024Representing graph data in a low-dimensional space for subsequent tasks is the purpose of attributed graph embedding. Most existing neural network approaches learn latent representations by minimizing reconstruction errors. Rare work considers the data distribution and the topological structure of latent codes simultaneously, which often results in inferior embeddings in real-world graph data. This paper proposes a novel Deep Manifold (Variational) Graph Auto-Encoder (DMVGAE/DMGAE) method for attributed graph data to improve the stability and quality of learned representations to tackle the crowding problem. The node-to-node geodesic similarity is preserved between the original and latent space under a pre-defined distribution. The proposed method surpasses state-of-the-art baseline algorithms by a significant margin on different downstream tasks across popular datasets, which validates our solutions. We promise to release the code after acceptance.
* This work has been accepted by ICASSP2023, due to download limitations, we upload this work here
MMDesign: Multi-Modality Transfer Learning for Generative Protein Design
Dec 11, 2023



Protein design involves generating protein sequences based on their corresponding protein backbones. While deep generative models show promise for learning protein design directly from data, the lack of publicly available structure-sequence pairings limits their generalization capabilities. Previous efforts of generative protein design have focused on architectural improvements and pseudo-data augmentation to overcome this bottleneck. To further address this challenge, we propose a novel protein design paradigm called MMDesign, which leverages multi-modality transfer learning. To our knowledge, MMDesign is the first framework that combines a pretrained structural module with a pretrained contextual module, using an auto-encoder (AE) based language model to incorporate prior semantic knowledge of protein sequences. We also introduce a cross-layer cross-modal alignment algorithm to enable the structural module to learn long-term temporal information and ensure consistency between structural and contextual modalities. Experimental results, only training with the small CATH dataset, demonstrate that our MMDesign framework consistently outperforms other baselines on various public test sets. To further assess the biological plausibility of the generated protein sequences and data distribution, we present systematic quantitative analysis techniques that provide interpretability and reveal more about the laws of protein design.
Segment Anything in Defect Detection
Nov 17, 2023



Defect detection plays a crucial role in infrared non-destructive testing systems, offering non-contact, safe, and efficient inspection capabilities. However, challenges such as low resolution, high noise, and uneven heating in infrared thermal images hinder comprehensive and accurate defect detection. In this study, we propose DefectSAM, a novel approach for segmenting defects on highly noisy thermal images based on the widely adopted model, Segment Anything (SAM)\cite{kirillov2023segany}. Harnessing the power of a meticulously curated dataset generated through labor-intensive lab experiments and valuable prompts from experienced experts, DefectSAM surpasses existing state-of-the-art segmentation algorithms and achieves significant improvements in defect detection rates. Notably, DefectSAM excels in detecting weaker and smaller defects on complex and irregular surfaces, reducing the occurrence of missed detections and providing more accurate defect size estimations. Experimental studies conducted on various materials have validated the effectiveness of our solutions in defect detection, which hold significant potential to expedite the evolution of defect detection tools, enabling enhanced inspection capabilities and accuracy in defect identification.
Lightweight Contrastive Protein Structure-Sequence Transformation
Mar 19, 2023



Pretrained protein structure models without labels are crucial foundations for the majority of protein downstream applications. The conventional structure pretraining methods follow the mature natural language pretraining methods such as denoised reconstruction and masked language modeling but usually destroy the real representation of spatial structures. The other common pretraining methods might predict a fixed set of predetermined object categories, where a restricted supervised manner limits their generality and usability as additional labeled data is required to specify any other protein concepts. In this work, we introduce a novel unsupervised protein structure representation pretraining with a robust protein language model. In particular, we first propose to leverage an existing pretrained language model to guide structure model learning through an unsupervised contrastive alignment. In addition, a self-supervised structure constraint is proposed to further learn the intrinsic information about the structures. With only light training data, the pretrained structure model can obtain better generalization ability. To quantitatively evaluate the proposed structure models, we design a series of rational evaluation methods, including internal tasks (e.g., contact map prediction, distribution alignment quality) and external/downstream tasks (e.g., protein design). The extensive experimental results conducted on multiple tasks and specific datasets demonstrate the superiority of the proposed sequence-structure transformation framework.
Protein Language Models and Structure Prediction: Connection and Progression
Nov 30, 2022



The prediction of protein structures from sequences is an important task for function prediction, drug design, and related biological processes understanding. Recent advances have proved the power of language models (LMs) in processing the protein sequence databases, which inherit the advantages of attention networks and capture useful information in learning representations for proteins. The past two years have witnessed remarkable success in tertiary protein structure prediction (PSP), including evolution-based and single-sequence-based PSP. It seems that instead of using energy-based models and sampling procedures, protein language model (pLM)-based pipelines have emerged as mainstream paradigms in PSP. Despite the fruitful progress, the PSP community needs a systematic and up-to-date survey to help bridge the gap between LMs in the natural language processing (NLP) and PSP domains and introduce their methodologies, advancements and practical applications. To this end, in this paper, we first introduce the similarities between protein and human languages that allow LMs extended to pLMs, and applied to protein databases. Then, we systematically review recent advances in LMs and pLMs from the perspectives of network architectures, pre-training strategies, applications, and commonly-used protein databases. Next, different types of methods for PSP are discussed, particularly how the pLM-based architectures function in the process of protein folding. Finally, we identify challenges faced by the PSP community and foresee promising research directions along with the advances of pLMs. This survey aims to be a hands-on guide for researchers to understand PSP methods, develop pLMs and tackle challenging problems in this field for practical purposes.
SimGRACE: A Simple Framework for Graph Contrastive Learning without Data Augmentation
Feb 11, 2022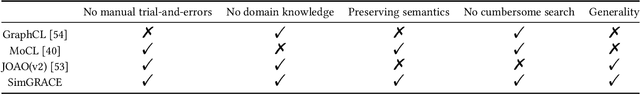

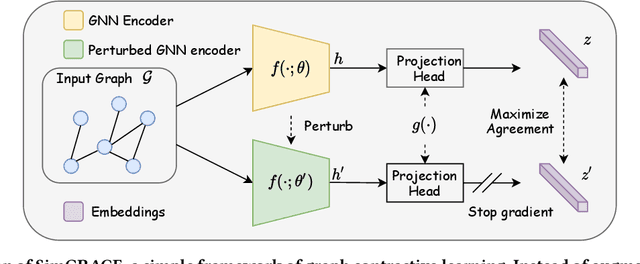
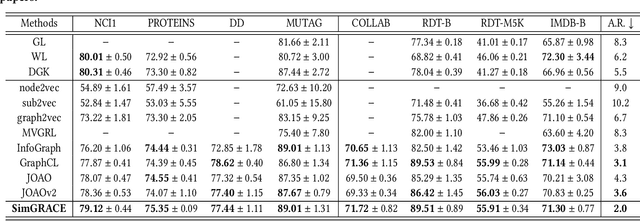
Graph contrastive learning (GCL) has emerged as a dominant technique for graph representation learning which maximizes the mutual information between paired graph augmentations that share the same semantics. Unfortunately, it is difficult to preserve semantics well during augmentations in view of the diverse nature of graph data. Currently, data augmentations in GCL that are designed to preserve semantics broadly fall into three unsatisfactory ways. First, the augmentations can be manually picked per dataset by trial-and-errors. Second, the augmentations can be selected via cumbersome search. Third, the augmentations can be obtained by introducing expensive domain-specific knowledge as guidance. All of these limit the efficiency and more general applicability of existing GCL methods. To circumvent these crucial issues, we propose a \underline{Sim}ple framework for \underline{GRA}ph \underline{C}ontrastive l\underline{E}arning, \textbf{SimGRACE} for brevity, which does not require data augmentations. Specifically, we take original graph as input and GNN model with its perturbed version as two encoders to obtain two correlated views for contrast. SimGRACE is inspired by the observation that graph data can preserve their semantics well during encoder perturbations while not requiring manual trial-and-errors, cumbersome search or expensive domain knowledge for augmentations selection. Also, we explain why SimGRACE can succeed. Furthermore, we devise adversarial training scheme, dubbed \textbf{AT-SimGRACE}, to enhance the robustness of graph contrastive learning and theoretically explain the reasons. Albeit simple, we show that SimGRACE can yield competitive or better performance compared with state-of-the-art methods in terms of generalizability, transferability and robustness, while enjoying unprecedented degree of flexibility and efficiency.
* Accepted by The Web Conference 2022 (WWW 2022)