 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTokenizing Electron Cloud in Protein-Ligand Interaction Learning
May 25, 2025



The affinity and specificity of protein-molecule binding directly impact functional outcomes, uncovering the mechanisms underlying biological regulation and signal transduction. Most deep-learning-based prediction approaches focus on structures of atoms or fragments. However, quantum chemical properties, such as electronic structures, are the key to unveiling interaction patterns but remain largely underexplored. To bridge this gap, we propose ECBind, a method for tokenizing electron cloud signals into quantized embeddings, enabling their integration into downstream tasks such as binding affinity prediction. By incorporating electron densities, ECBind helps uncover binding modes that cannot be fully represented by atom-level models. Specifically, to remove the redundancy inherent in electron cloud signals, a structure-aware transformer and hierarchical codebooks encode 3D binding sites enriched with electron structures into tokens. These tokenized codes are then used for specific tasks with labels. To extend its applicability to a wider range of scenarios, we utilize knowledge distillation to develop an electron-cloud-agnostic prediction model. Experimentally, ECBind demonstrates state-of-the-art performance across multiple tasks, achieving improvements of 6.42\% and 15.58\% in per-structure Pearson and Spearman correlation coefficients, respectively.
A Simple yet Effective DDG Predictor is An Unsupervised Antibody Optimizer and Explainer
Feb 10, 2025The proteins that exist today have been optimized over billions of years of natural evolution, during which nature creates random mutations and selects them. The discovery of functionally promising mutations is challenged by the limited evolutionary accessible regions, i.e., only a small region on the fitness landscape is beneficial. There have been numerous priors used to constrain protein evolution to regions of landscapes with high-fitness variants, among which the change in binding free energy (DDG) of protein complexes upon mutations is one of the most commonly used priors. However, the huge mutation space poses two challenges: (1) how to improve the efficiency of DDG prediction for fast mutation screening; and (2) how to explain mutation preferences and efficiently explore accessible evolutionary regions. To address these challenges, we propose a lightweight DDG predictor (Light-DDG), which adopts a structure-aware Transformer as the backbone and enhances it by knowledge distilled from existing powerful but computationally heavy DDG predictors. Additionally, we augmented, annotated, and released a large-scale dataset containing millions of mutation data for pre-training Light-DDG. We find that such a simple yet effective Light-DDG can serve as a good unsupervised antibody optimizer and explainer. For the target antibody, we propose a novel Mutation Explainer to learn mutation preferences, which accounts for the marginal benefit of each mutation per residue. To further explore accessible evolutionary regions, we conduct preference-guided antibody optimization and evaluate antibody candidates quickly using Light-DDG to identify desirable mutations.
FM-TS: Flow Matching for Time Series Generation
Nov 12, 2024
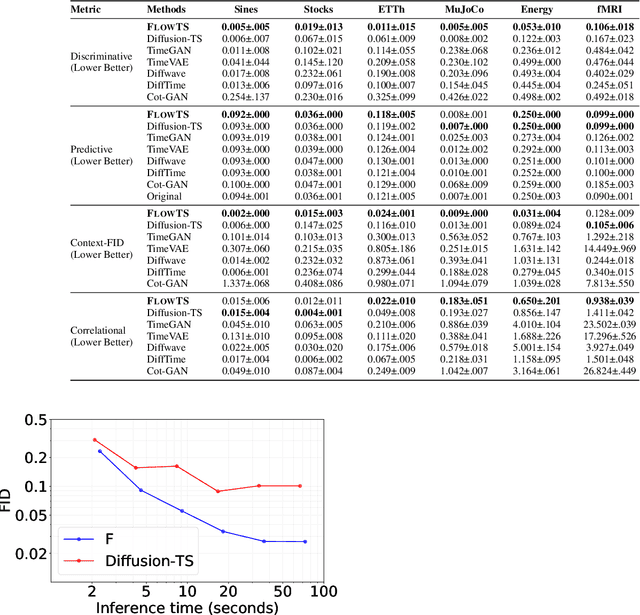


Time series generation has emerged as an essential tool for analyzing temporal data across numerous fields. While diffusion models have recently gained significant attention in generating high-quality time series, they tend to be computationally demanding and reliant on complex stochastic processes. To address these limitations, we introduce FM-TS, a rectified Flow Matching-based framework for Time Series generation, which simplifies the time series generation process by directly optimizing continuous trajectories. This approach avoids the need for iterative sampling or complex noise schedules typically required in diffusion-based models. FM-TS is more efficient in terms of training and inference. Moreover, FM-TS is highly adaptive, supporting both conditional and unconditional time series generation. Notably, through our novel inference design, the model trained in an unconditional setting can seamlessly generalize to conditional tasks without the need for retraining. Extensive benchmarking across both settings demonstrates that FM-TS consistently delivers superior performance compared to existing approaches while being more efficient in terms of training and inference. For instance, in terms of discriminative score, FM-TS achieves 0.005, 0.019, 0.011, 0.005, 0.053, and 0.106 on the Sines, Stocks, ETTh, MuJoCo, Energy, and fMRI unconditional time series datasets, respectively, significantly outperforming the second-best method which achieves 0.006, 0.067, 0.061, 0.008, 0.122, and 0.167 on the same datasets. We have achieved superior performance in solar forecasting and MuJoCo imputation tasks, significantly enhanced by our innovative $t$ power sampling method. The code is available at https://github.com/UNITES-Lab/FMTS.
MeToken: Uniform Micro-environment Token Boosts Post-Translational Modification Prediction
Nov 04, 2024



Post-translational modifications (PTMs) profoundly expand the complexity and functionality of the proteome, regulating protein attributes and interactions that are crucial for biological processes. Accurately predicting PTM sites and their specific types is therefore essential for elucidating protein function and understanding disease mechanisms. Existing computational approaches predominantly focus on protein sequences to predict PTM sites, driven by the recognition of sequence-dependent motifs. However, these approaches often overlook protein structural contexts. In this work, we first compile a large-scale sequence-structure PTM dataset, which serves as the foundation for fair comparison. We introduce the MeToken model, which tokenizes the micro-environment of each amino acid, integrating both sequence and structural information into unified discrete tokens. This model not only captures the typical sequence motifs associated with PTMs but also leverages the spatial arrangements dictated by protein tertiary structures, thus providing a holistic view of the factors influencing PTM sites. Designed to address the long-tail distribution of PTM types, MeToken employs uniform sub-codebooks that ensure even the rarest PTMs are adequately represented and distinguished. We validate the effectiveness and generalizability of MeToken across multiple datasets, demonstrating its superior performance in accurately identifying PTM types. The results underscore the importance of incorporating structural data and highlight MeToken's potential in facilitating accurate and comprehensive PTM predictions, which could significantly impact proteomics research. The code and datasets are available at https://github.com/A4Bio/MeToken.
Learning to Model Graph Structural Information on MLPs via Graph Structure Self-Contrasting
Sep 09, 2024



Recent years have witnessed great success in handling graph-related tasks with Graph Neural Networks (GNNs). However, most existing GNNs are based on message passing to perform feature aggregation and transformation, where the structural information is explicitly involved in the forward propagation by coupling with node features through graph convolution at each layer. As a result, subtle feature noise or structure perturbation may cause severe error propagation, resulting in extremely poor robustness. In this paper, we rethink the roles played by graph structural information in graph data training and identify that message passing is not the only path to modeling structural information. Inspired by this, we propose a simple but effective Graph Structure Self-Contrasting (GSSC) framework that learns graph structural information without message passing. The proposed framework is based purely on Multi-Layer Perceptrons (MLPs), where the structural information is only implicitly incorporated as prior knowledge to guide the computation of supervision signals, substituting the explicit message propagation as in GNNs. Specifically, it first applies structural sparsification to remove potentially uninformative or noisy edges in the neighborhood, and then performs structural self-contrasting in the sparsified neighborhood to learn robust node representations. Finally, structural sparsification and self-contrasting are formulated as a bi-level optimization problem and solved in a unified framework. Extensive experiments have qualitatively and quantitatively demonstrated that the GSSC framework can produce truly encouraging performance with better generalization and robustness than other leading competitors.
Teach Harder, Learn Poorer: Rethinking Hard Sample Distillation for GNN-to-MLP Knowledge Distillation
Jul 20, 2024


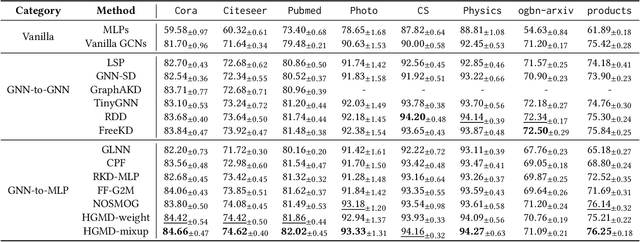
To bridge the gaps between powerful Graph Neural Networks (GNNs) and lightweight Multi-Layer Perceptron (MLPs), GNN-to-MLP Knowledge Distillation (KD) proposes to distill knowledge from a well-trained teacher GNN into a student MLP. In this paper, we revisit the knowledge samples (nodes) in teacher GNNs from the perspective of hardness, and identify that hard sample distillation may be a major performance bottleneck of existing graph KD algorithms. The GNN-to-MLP KD involves two different types of hardness, one student-free knowledge hardness describing the inherent complexity of GNN knowledge, and the other student-dependent distillation hardness describing the difficulty of teacher-to-student distillation. However, most of the existing work focuses on only one of these aspects or regards them as one thing. This paper proposes a simple yet effective Hardness-aware GNN-to-MLP Distillation (HGMD) framework, which decouples the two hardnesses and estimates them using a non-parametric approach. Finally, two hardness-aware distillation schemes (i.e., HGMD-weight and HGMD-mixup) are further proposed to distill hardness-aware knowledge from teacher GNNs into the corresponding nodes of student MLPs. As non-parametric distillation, HGMD does not involve any additional learnable parameters beyond the student MLPs, but it still outperforms most of the state-of-the-art competitors. HGMD-mixup improves over the vanilla MLPs by 12.95% and outperforms its teacher GNNs by 2.48% averaged over seven real-world datasets.
The Heterophilic Graph Learning Handbook: Benchmarks, Models, Theoretical Analysis, Applications and Challenges
Jul 12, 2024



Homophily principle, \ie{} nodes with the same labels or similar attributes are more likely to be connected, has been commonly believed to be the main reason for the superiority of Graph Neural Networks (GNNs) over traditional Neural Networks (NNs) on graph-structured data, especially on node-level tasks. However, recent work has identified a non-trivial set of datasets where GNN's performance compared to the NN's is not satisfactory. Heterophily, i.e. low homophily, has been considered the main cause of this empirical observation. People have begun to revisit and re-evaluate most existing graph models, including graph transformer and its variants, in the heterophily scenario across various kinds of graphs, e.g. heterogeneous graphs, temporal graphs and hypergraphs. Moreover, numerous graph-related applications are found to be closely related to the heterophily problem. In the past few years, considerable effort has been devoted to studying and addressing the heterophily issue. In this survey, we provide a comprehensive review of the latest progress on heterophilic graph learning, including an extensive summary of benchmark datasets and evaluation of homophily metrics on synthetic graphs, meticulous classification of the most updated supervised and unsupervised learning methods, thorough digestion of the theoretical analysis on homophily/heterophily, and broad exploration of the heterophily-related applications. Notably, through detailed experiments, we are the first to categorize benchmark heterophilic datasets into three sub-categories: malignant, benign and ambiguous heterophily. Malignant and ambiguous datasets are identified as the real challenging datasets to test the effectiveness of new models on the heterophily challenge. Finally, we propose several challenges and future directions for heterophilic graph representation learning.
CBGBench: Fill in the Blank of Protein-Molecule Complex Binding Graph
Jun 16, 2024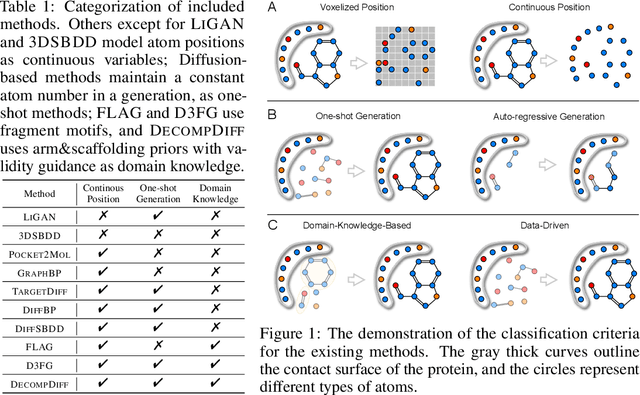



Structure-based drug design (SBDD) aims to generate potential drugs that can bind to a target protein and is greatly expedited by the aid of AI techniques in generative models. However, a lack of systematic understanding persists due to the diverse settings, complex implementation, difficult reproducibility, and task singularity. Firstly, the absence of standardization can lead to unfair comparisons and inconclusive insights. To address this dilemma, we propose CBGBench, a comprehensive benchmark for SBDD, that unifies the task as a generative heterogeneous graph completion, analogous to fill-in-the-blank of the 3D complex binding graph. By categorizing existing methods based on their attributes, CBGBench facilitates a modular and extensible framework that implements various cutting-edge methods. Secondly, a single task on \textit{de novo} molecule generation can hardly reflect their capabilities. To broaden the scope, we have adapted these models to a range of tasks essential in drug design, which are considered sub-tasks within the graph fill-in-the-blank tasks. These tasks include the generative designation of \textit{de novo} molecules, linkers, fragments, scaffolds, and sidechains, all conditioned on the structures of protein pockets. Our evaluations are conducted with fairness, encompassing comprehensive perspectives on interaction, chemical properties, geometry authenticity, and substructure validity. We further provide the pre-trained versions of the state-of-the-art models and deep insights with analysis from empirical studies. The codebase for CBGBench is publicly accessible at \url{https://github.com/Edapinenut/CBGBench}.
UniIF: Unified Molecule Inverse Folding
May 29, 2024
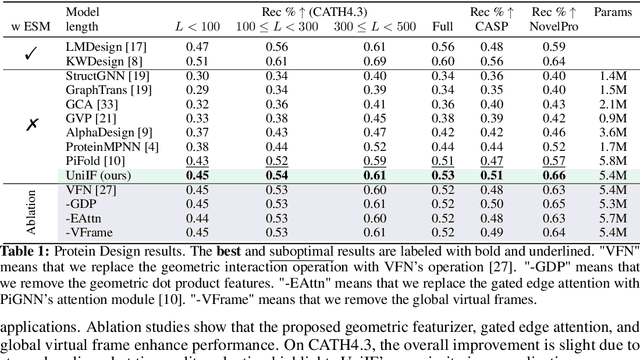
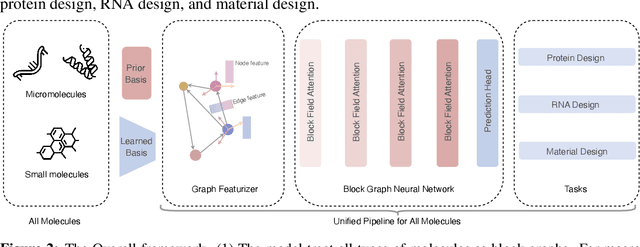

Molecule inverse folding has been a long-standing challenge in chemistry and biology, with the potential to revolutionize drug discovery and material science. Despite specified models have been proposed for different small- or macro-molecules, few have attempted to unify the learning process, resulting in redundant efforts. Complementary to recent advancements in molecular structure prediction, such as RoseTTAFold All-Atom and AlphaFold3, we propose the unified model UniIF for the inverse folding of all molecules. We do such unification in two levels: 1) Data-Level: We propose a unified block graph data form for all molecules, including the local frame building and geometric feature initialization. 2) Model-Level: We introduce a geometric block attention network, comprising a geometric interaction, interactive attention and virtual long-term dependency modules, to capture the 3D interactions of all molecules. Through comprehensive evaluations across various tasks such as protein design, RNA design, and material design, we demonstrate that our proposed method surpasses state-of-the-art methods on all tasks. UniIF offers a versatile and effective solution for general molecule inverse folding.
Learning to Predict Mutation Effects of Protein-Protein Interactions by Microenvironment-aware Hierarchical Prompt Learning
May 16, 2024Protein-protein bindings play a key role in a variety of fundamental biological processes, and thus predicting the effects of amino acid mutations on protein-protein binding is crucial. To tackle the scarcity of annotated mutation data, pre-training with massive unlabeled data has emerged as a promising solution. However, this process faces a series of challenges: (1) complex higher-order dependencies among multiple (more than paired) structural scales have not yet been fully captured; (2) it is rarely explored how mutations alter the local conformation of the surrounding microenvironment; (3) pre-training is costly, both in data size and computational burden. In this paper, we first construct a hierarchical prompt codebook to record common microenvironmental patterns at different structural scales independently. Then, we develop a novel codebook pre-training task, namely masked microenvironment modeling, to model the joint distribution of each mutation with their residue types, angular statistics, and local conformational changes in the microenvironment. With the constructed prompt codebook, we encode the microenvironment around each mutation into multiple hierarchical prompts and combine them to flexibly provide information to wild-type and mutated protein complexes about their microenvironmental differences. Such a hierarchical prompt learning framework has demonstrated superior performance and training efficiency over state-of-the-art pre-training-based methods in mutation effect prediction and a case study of optimizing human antibodies against SARS-CoV-2.