 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeReGuLaR: Variational Latent Reasoning Guided by Rendered Chain-of-Thought
Jan 30, 2026While Chain-of-Thought (CoT) significantly enhances the performance of Large Language Models (LLMs), explicit reasoning chains introduce substantial computational redundancy. Recent latent reasoning methods attempt to mitigate this by compressing reasoning processes into latent space, but often suffer from severe performance degradation due to the lack of appropriate compression guidance. In this study, we propose Rendered CoT-Guided variational Latent Reasoning (ReGuLaR), a simple yet novel latent learning paradigm resolving this issue. Fundamentally, we formulate latent reasoning within the Variational Auto-Encoding (VAE) framework, sampling the current latent reasoning state from the posterior distribution conditioned on previous ones. Specifically, when learning this variational latent reasoning model, we render explicit reasoning chains as images, from which we extract dense visual-semantic representations to regularize the posterior distribution, thereby achieving efficient compression with minimal information loss. Extensive experiments demonstrate that ReGuLaR significantly outperforms existing latent reasoning methods across both computational efficiency and reasoning effectiveness, and even surpasses CoT through multi-modal reasoning, providing a new and insightful solution to latent reasoning. Code: https://github.com/FanmengWang/ReGuLaR.
Innovator-VL: A Multimodal Large Language Model for Scientific Discovery
Jan 27, 2026We present Innovator-VL, a scientific multimodal large language model designed to advance understanding and reasoning across diverse scientific domains while maintaining excellent performance on general vision tasks. Contrary to the trend of relying on massive domain-specific pretraining and opaque pipelines, our work demonstrates that principled training design and transparent methodology can yield strong scientific intelligence with substantially reduced data requirements. (i) First, we provide a fully transparent, end-to-end reproducible training pipeline, covering data collection, cleaning, preprocessing, supervised fine-tuning, reinforcement learning, and evaluation, along with detailed optimization recipes. This facilitates systematic extension by the community. (ii) Second, Innovator-VL exhibits remarkable data efficiency, achieving competitive performance on various scientific tasks using fewer than five million curated samples without large-scale pretraining. These results highlight that effective reasoning can be achieved through principled data selection rather than indiscriminate scaling. (iii) Third, Innovator-VL demonstrates strong generalization, achieving competitive performance on general vision, multimodal reasoning, and scientific benchmarks. This indicates that scientific alignment can be integrated into a unified model without compromising general-purpose capabilities. Our practices suggest that efficient, reproducible, and high-performing scientific multimodal models can be built even without large-scale data, providing a practical foundation for future research.
T$^\star$: Progressive Block Scaling for MDM Through Trajectory Aware RL
Jan 16, 2026We present T$^\star$, a simple \textsc{TraceRL}-based training curriculum for progressive block-size scaling in masked diffusion language models (MDMs). Starting from an AR-initialized small-block MDM, T$^\star$~transitions smoothly to larger blocks, enabling higher-parallelism decoding with minimal performance degradation on math reasoning benchmarks. Moreover, further analysis suggests that T$^\star$~can converge to an alternative decoding schedule $\hat{\rm S}$ that achieves comparable performance.
A Simple yet Effective DDG Predictor is An Unsupervised Antibody Optimizer and Explainer
Feb 10, 2025The proteins that exist today have been optimized over billions of years of natural evolution, during which nature creates random mutations and selects them. The discovery of functionally promising mutations is challenged by the limited evolutionary accessible regions, i.e., only a small region on the fitness landscape is beneficial. There have been numerous priors used to constrain protein evolution to regions of landscapes with high-fitness variants, among which the change in binding free energy (DDG) of protein complexes upon mutations is one of the most commonly used priors. However, the huge mutation space poses two challenges: (1) how to improve the efficiency of DDG prediction for fast mutation screening; and (2) how to explain mutation preferences and efficiently explore accessible evolutionary regions. To address these challenges, we propose a lightweight DDG predictor (Light-DDG), which adopts a structure-aware Transformer as the backbone and enhances it by knowledge distilled from existing powerful but computationally heavy DDG predictors. Additionally, we augmented, annotated, and released a large-scale dataset containing millions of mutation data for pre-training Light-DDG. We find that such a simple yet effective Light-DDG can serve as a good unsupervised antibody optimizer and explainer. For the target antibody, we propose a novel Mutation Explainer to learn mutation preferences, which accounts for the marginal benefit of each mutation per residue. To further explore accessible evolutionary regions, we conduct preference-guided antibody optimization and evaluate antibody candidates quickly using Light-DDG to identify desirable mutations.
Intelligent System for Automated Molecular Patent Infringement Assessment
Dec 10, 2024



Automated drug discovery offers significant potential for accelerating the development of novel therapeutics by substituting labor-intensive human workflows with machine-driven processes. However, a critical bottleneck persists in the inability of current automated frameworks to assess whether newly designed molecules infringe upon existing patents, posing significant legal and financial risks. We introduce PatentFinder, a novel tool-enhanced and multi-agent framework that accurately and comprehensively evaluates small molecules for patent infringement. It incorporates both heuristic and model-based tools tailored for decomposed subtasks, featuring: MarkushParser, which is capable of optical chemical structure recognition of molecular and Markush structures, and MarkushMatcher, which enhances large language models' ability to extract substituent groups from molecules accurately. On our benchmark dataset MolPatent-240, PatentFinder outperforms baseline approaches that rely solely on large language models, demonstrating a 13.8\% increase in F1-score and a 12\% rise in accuracy. Experimental results demonstrate that PatentFinder mitigates label bias to produce balanced predictions and autonomously generates detailed, interpretable patent infringement reports. This work not only addresses a pivotal challenge in automated drug discovery but also demonstrates the potential of decomposing complex scientific tasks into manageable subtasks for specialized, tool-augmented agents.
Learning to Model Graph Structural Information on MLPs via Graph Structure Self-Contrasting
Sep 09, 2024



Recent years have witnessed great success in handling graph-related tasks with Graph Neural Networks (GNNs). However, most existing GNNs are based on message passing to perform feature aggregation and transformation, where the structural information is explicitly involved in the forward propagation by coupling with node features through graph convolution at each layer. As a result, subtle feature noise or structure perturbation may cause severe error propagation, resulting in extremely poor robustness. In this paper, we rethink the roles played by graph structural information in graph data training and identify that message passing is not the only path to modeling structural information. Inspired by this, we propose a simple but effective Graph Structure Self-Contrasting (GSSC) framework that learns graph structural information without message passing. The proposed framework is based purely on Multi-Layer Perceptrons (MLPs), where the structural information is only implicitly incorporated as prior knowledge to guide the computation of supervision signals, substituting the explicit message propagation as in GNNs. Specifically, it first applies structural sparsification to remove potentially uninformative or noisy edges in the neighborhood, and then performs structural self-contrasting in the sparsified neighborhood to learn robust node representations. Finally, structural sparsification and self-contrasting are formulated as a bi-level optimization problem and solved in a unified framework. Extensive experiments have qualitatively and quantitatively demonstrated that the GSSC framework can produce truly encouraging performance with better generalization and robustness than other leading competitors.
CBGBench: Fill in the Blank of Protein-Molecule Complex Binding Graph
Jun 16, 2024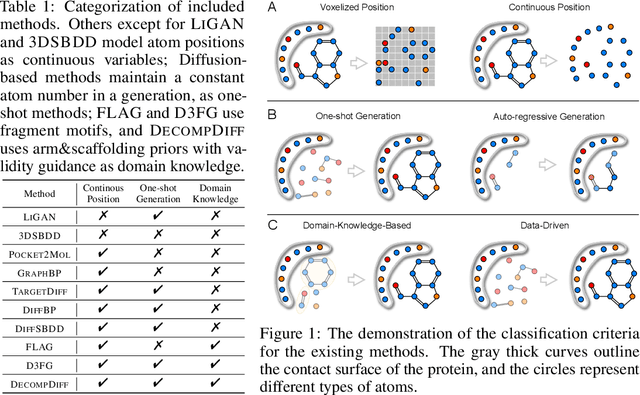



Structure-based drug design (SBDD) aims to generate potential drugs that can bind to a target protein and is greatly expedited by the aid of AI techniques in generative models. However, a lack of systematic understanding persists due to the diverse settings, complex implementation, difficult reproducibility, and task singularity. Firstly, the absence of standardization can lead to unfair comparisons and inconclusive insights. To address this dilemma, we propose CBGBench, a comprehensive benchmark for SBDD, that unifies the task as a generative heterogeneous graph completion, analogous to fill-in-the-blank of the 3D complex binding graph. By categorizing existing methods based on their attributes, CBGBench facilitates a modular and extensible framework that implements various cutting-edge methods. Secondly, a single task on \textit{de novo} molecule generation can hardly reflect their capabilities. To broaden the scope, we have adapted these models to a range of tasks essential in drug design, which are considered sub-tasks within the graph fill-in-the-blank tasks. These tasks include the generative designation of \textit{de novo} molecules, linkers, fragments, scaffolds, and sidechains, all conditioned on the structures of protein pockets. Our evaluations are conducted with fairness, encompassing comprehensive perspectives on interaction, chemical properties, geometry authenticity, and substructure validity. We further provide the pre-trained versions of the state-of-the-art models and deep insights with analysis from empirical studies. The codebase for CBGBench is publicly accessible at \url{https://github.com/Edapinenut/CBGBench}.
A Teacher-Free Graph Knowledge Distillation Framework with Dual Self-Distillation
Mar 06, 2024



Recent years have witnessed great success in handling graph-related tasks with Graph Neural Networks (GNNs). Despite their great academic success, Multi-Layer Perceptrons (MLPs) remain the primary workhorse for practical industrial applications. One reason for such an academic-industry gap is the neighborhood-fetching latency incurred by data dependency in GNNs. To reduce their gaps, Graph Knowledge Distillation (GKD) is proposed, usually based on a standard teacher-student architecture, to distill knowledge from a large teacher GNN into a lightweight student GNN or MLP. However, we found in this paper that neither teachers nor GNNs are necessary for graph knowledge distillation. We propose a Teacher-Free Graph Self-Distillation (TGS) framework that does not require any teacher model or GNNs during both training and inference. More importantly, the proposed TGS framework is purely based on MLPs, where structural information is only implicitly used to guide dual knowledge self-distillation between the target node and its neighborhood. As a result, TGS enjoys the benefits of graph topology awareness in training but is free from data dependency in inference. Extensive experiments have shown that the performance of vanilla MLPs can be greatly improved with dual self-distillation, e.g., TGS improves over vanilla MLPs by 15.54% on average and outperforms state-of-the-art GKD algorithms on six real-world datasets. In terms of inference speed, TGS infers 75X-89X faster than existing GNNs and 16X-25X faster than classical inference acceleration methods.
SQL-CRAFT: Text-to-SQL through Interactive Refinement and Enhanced Reasoning
Feb 20, 2024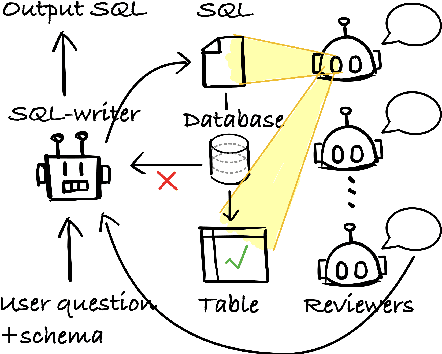
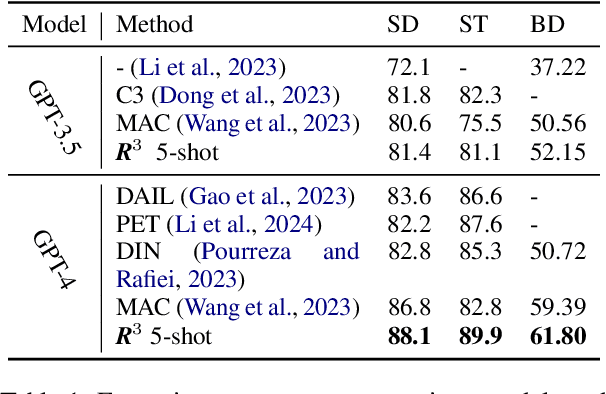
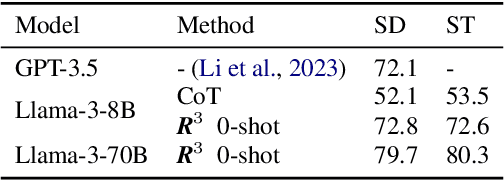
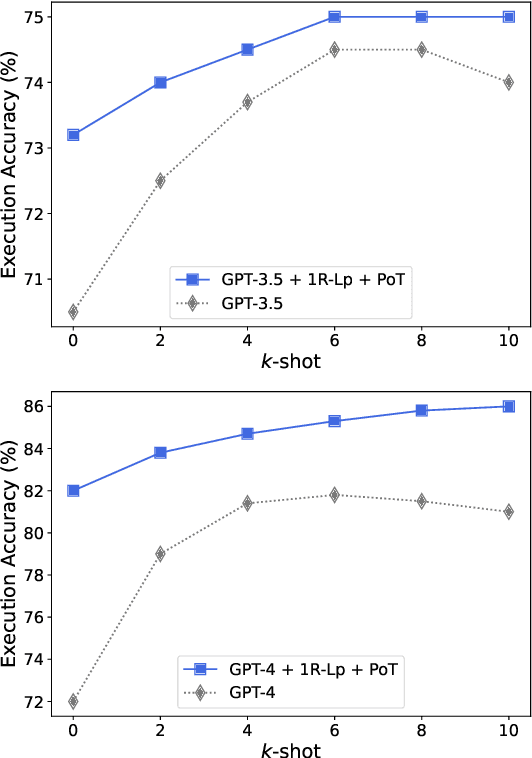
Modern LLMs have become increasingly powerful, but they are still facing challenges in specialized tasks such as Text-to-SQL. We propose SQL-CRAFT, a framework to advance LLMs' SQL generation Capabilities through inteRActive reFinemenT and enhanced reasoning. We leverage an Interactive Correction Loop (IC-Loop) for LLMs to interact with databases automatically, as well as Python-enhanced reasoning. We conduct experiments on two Text-to-SQL datasets, Spider and Bird, with performance improvements of up to 5.7% compared to the naive prompting method. Moreover, our method surpasses the current state-of-the-art on the Spider Leaderboard, demonstrating the effectiveness of our framework.
Uni-QSAR: an Auto-ML Tool for Molecular Property Prediction
Apr 24, 2023



Recently deep learning based quantitative structure-activity relationship (QSAR) models has shown surpassing performance than traditional methods for property prediction tasks in drug discovery. However, most DL based QSAR models are restricted to limited labeled data to achieve better performance, and also are sensitive to model scale and hyper-parameters. In this paper, we propose Uni-QSAR, a powerful Auto-ML tool for molecule property prediction tasks. Uni-QSAR combines molecular representation learning (MRL) of 1D sequential tokens, 2D topology graphs, and 3D conformers with pretraining models to leverage rich representation from large-scale unlabeled data. Without any manual fine-tuning or model selection, Uni-QSAR outperforms SOTA in 21/22 tasks of the Therapeutic Data Commons (TDC) benchmark under designed parallel workflow, with an average performance improvement of 6.09\%. Furthermore, we demonstrate the practical usefulness of Uni-QSAR in drug discovery domains.