 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMemOCR: Layout-Aware Visual Memory for Efficient Long-Horizon Reasoning
Jan 29, 2026Long-horizon agentic reasoning necessitates effectively compressing growing interaction histories into a limited context window. Most existing memory systems serialize history as text, where token-level cost is uniform and scales linearly with length, often spending scarce budget on low-value details. To this end, we introduce MemOCR, a multimodal memory agent that improves long-horizon reasoning under tight context budgets by allocating memory space with adaptive information density through visual layout. Concretely, MemOCR maintains a structured rich-text memory (e.g., headings, highlights) and renders it into an image that the agent consults for memory access, visually prioritizing crucial evidence while aggressively compressing auxiliary details. To ensure robustness across varying memory budgets, we train MemOCR with reinforcement learning under budget-aware objectives that expose the agent to diverse compression levels. Across long-context multi-hop and single-hop question-answering benchmarks, MemOCR outperforms strong text-based baselines and achieves more effective context utilization under extreme budgets.
LongCat-Flash-Thinking-2601 Technical Report
Jan 23, 2026We introduce LongCat-Flash-Thinking-2601, a 560-billion-parameter open-source Mixture-of-Experts (MoE) reasoning model with superior agentic reasoning capability. LongCat-Flash-Thinking-2601 achieves state-of-the-art performance among open-source models on a wide range of agentic benchmarks, including agentic search, agentic tool use, and tool-integrated reasoning. Beyond benchmark performance, the model demonstrates strong generalization to complex tool interactions and robust behavior under noisy real-world environments. Its advanced capability stems from a unified training framework that combines domain-parallel expert training with subsequent fusion, together with an end-to-end co-design of data construction, environments, algorithms, and infrastructure spanning from pre-training to post-training. In particular, the model's strong generalization capability in complex tool-use are driven by our in-depth exploration of environment scaling and principled task construction. To optimize long-tailed, skewed generation and multi-turn agentic interactions, and to enable stable training across over 10,000 environments spanning more than 20 domains, we systematically extend our asynchronous reinforcement learning framework, DORA, for stable and efficient large-scale multi-environment training. Furthermore, recognizing that real-world tasks are inherently noisy, we conduct a systematic analysis and decomposition of real-world noise patterns, and design targeted training procedures to explicitly incorporate such imperfections into the training process, resulting in improved robustness for real-world applications. To further enhance performance on complex reasoning tasks, we introduce a Heavy Thinking mode that enables effective test-time scaling by jointly expanding reasoning depth and width through intensive parallel thinking.
Search and Refine During Think: Autonomous Retrieval-Augmented Reasoning of LLMs
May 16, 2025Large language models have demonstrated impressive reasoning capabilities but are inherently limited by their knowledge reservoir. Retrieval-augmented reasoning mitigates this limitation by allowing LLMs to query external resources, but existing methods often retrieve irrelevant or noisy information, hindering accurate reasoning. In this paper, we propose AutoRefine, a reinforcement learning post-training framework that adopts a new ``search-and-refine-during-think'' paradigm. AutoRefine introduces explicit knowledge refinement steps between successive search calls, enabling the model to iteratively filter, distill, and organize evidence before generating an answer. Furthermore, we incorporate tailored retrieval-specific rewards alongside answer correctness rewards using group relative policy optimization. Experiments on single-hop and multi-hop QA benchmarks demonstrate that AutoRefine significantly outperforms existing approaches, particularly in complex, multi-hop reasoning scenarios. Detailed analysis shows that AutoRefine issues frequent, higher-quality searches and synthesizes evidence effectively.
Multimodal Language Modeling for High-Accuracy Single Cell Transcriptomics Analysis and Generation
Mar 12, 2025Pre-trained language models (PLMs) have revolutionized scientific research, yet their application to single-cell analysis remains limited. Text PLMs cannot process single-cell RNA sequencing data, while cell PLMs lack the ability to handle free text, restricting their use in multimodal tasks. Existing efforts to bridge these modalities often suffer from information loss or inadequate single-modal pre-training, leading to suboptimal performances. To address these challenges, we propose Single-Cell MultiModal Generative Pre-trained Transformer (scMMGPT), a unified PLM for joint cell and text modeling. scMMGPT effectively integrates the state-of-the-art cell and text PLMs, facilitating cross-modal knowledge sharing for improved performance. To bridge the text-cell modality gap, scMMGPT leverages dedicated cross-modal projectors, and undergoes extensive pre-training on 27 million cells -- the largest dataset for multimodal cell-text PLMs to date. This large-scale pre-training enables scMMGPT to excel in joint cell-text tasks, achieving an 84\% relative improvement of textual discrepancy for cell description generation, 20.5\% higher accuracy for cell type annotation, and 4\% improvement in $k$-NN accuracy for text-conditioned pseudo-cell generation, outperforming baselines.
NExT-Mol: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation
Feb 18, 20253D molecule generation is crucial for drug discovery and material design. While prior efforts focus on 3D diffusion models for their benefits in modeling continuous 3D conformers, they overlook the advantages of 1D SELFIES-based Language Models (LMs), which can generate 100% valid molecules and leverage the billion-scale 1D molecule datasets. To combine these advantages for 3D molecule generation, we propose a foundation model -- NExT-Mol: 3D Diffusion Meets 1D Language Modeling for 3D Molecule Generation. NExT-Mol uses an extensively pretrained molecule LM for 1D molecule generation, and subsequently predicts the generated molecule's 3D conformers with a 3D diffusion model. We enhance NExT-Mol's performance by scaling up the LM's model size, refining the diffusion neural architecture, and applying 1D to 3D transfer learning. Notably, our 1D molecule LM significantly outperforms baselines in distributional similarity while ensuring validity, and our 3D diffusion model achieves leading performances in conformer prediction. Given these improvements in 1D and 3D modeling, NExT-Mol achieves a 26% relative improvement in 3D FCD for de novo 3D generation on GEOM-DRUGS, and a 13% average relative gain for conditional 3D generation on QM9-2014. Our codes and pretrained checkpoints are available at https://github.com/acharkq/NExT-Mol.
Intelligent System for Automated Molecular Patent Infringement Assessment
Dec 10, 2024



Automated drug discovery offers significant potential for accelerating the development of novel therapeutics by substituting labor-intensive human workflows with machine-driven processes. However, a critical bottleneck persists in the inability of current automated frameworks to assess whether newly designed molecules infringe upon existing patents, posing significant legal and financial risks. We introduce PatentFinder, a novel tool-enhanced and multi-agent framework that accurately and comprehensively evaluates small molecules for patent infringement. It incorporates both heuristic and model-based tools tailored for decomposed subtasks, featuring: MarkushParser, which is capable of optical chemical structure recognition of molecular and Markush structures, and MarkushMatcher, which enhances large language models' ability to extract substituent groups from molecules accurately. On our benchmark dataset MolPatent-240, PatentFinder outperforms baseline approaches that rely solely on large language models, demonstrating a 13.8\% increase in F1-score and a 12\% rise in accuracy. Experimental results demonstrate that PatentFinder mitigates label bias to produce balanced predictions and autonomously generates detailed, interpretable patent infringement reports. This work not only addresses a pivotal challenge in automated drug discovery but also demonstrates the potential of decomposing complex scientific tasks into manageable subtasks for specialized, tool-augmented agents.
SciLitLLM: How to Adapt LLMs for Scientific Literature Understanding
Aug 30, 2024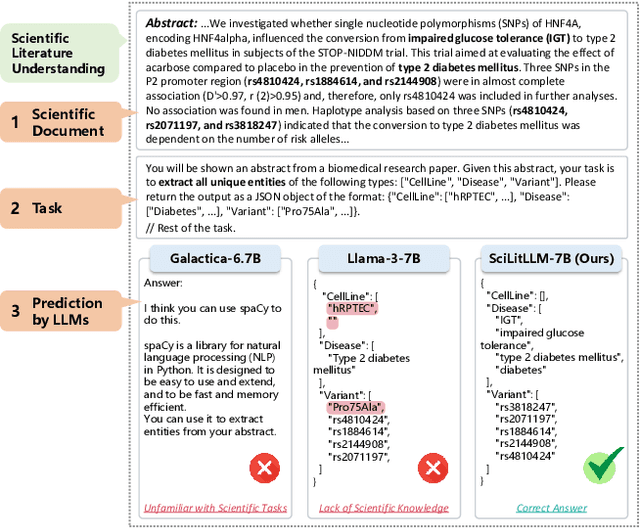
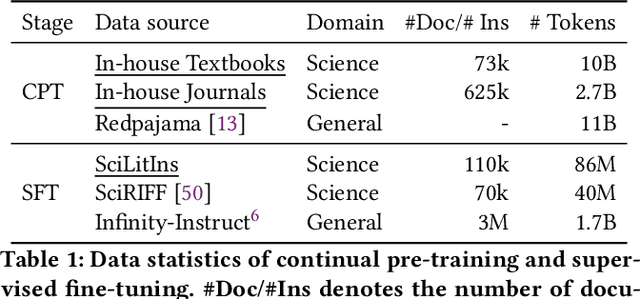
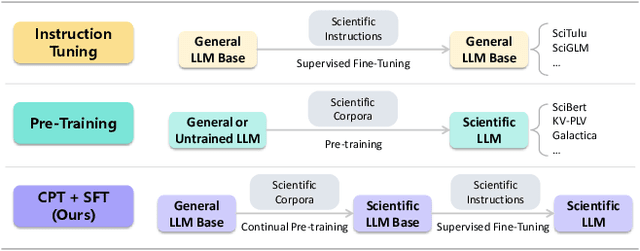
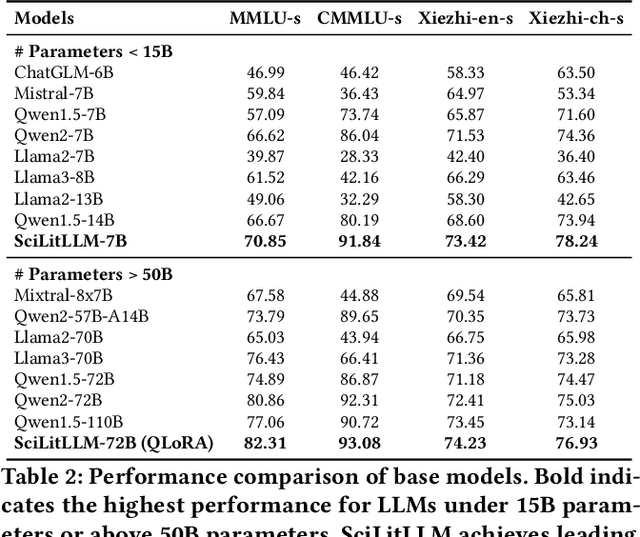
Scientific literature understanding is crucial for extracting targeted information and garnering insights, thereby significantly advancing scientific discovery. Despite the remarkable success of Large Language Models (LLMs), they face challenges in scientific literature understanding, primarily due to (1) a lack of scientific knowledge and (2) unfamiliarity with specialized scientific tasks. To develop an LLM specialized in scientific literature understanding, we propose a hybrid strategy that integrates continual pre-training (CPT) and supervised fine-tuning (SFT), to simultaneously infuse scientific domain knowledge and enhance instruction-following capabilities for domain-specific tasks.cIn this process, we identify two key challenges: (1) constructing high-quality CPT corpora, and (2) generating diverse SFT instructions. We address these challenges through a meticulous pipeline, including PDF text extraction, parsing content error correction, quality filtering, and synthetic instruction creation. Applying this strategy, we present a suite of LLMs: SciLitLLM, specialized in scientific literature understanding. These models demonstrate promising performance on scientific literature understanding benchmarks. Our contributions are threefold: (1) We present an effective framework that integrates CPT and SFT to adapt LLMs to scientific literature understanding, which can also be easily adapted to other domains. (2) We propose an LLM-based synthesis method to generate diverse and high-quality scientific instructions, resulting in a new instruction set -- SciLitIns -- for supervised fine-tuning in less-represented scientific domains. (3) SciLitLLM achieves promising performance improvements on scientific literature understanding benchmarks.
ReactXT: Understanding Molecular "Reaction-ship" via Reaction-Contextualized Molecule-Text Pretraining
May 23, 2024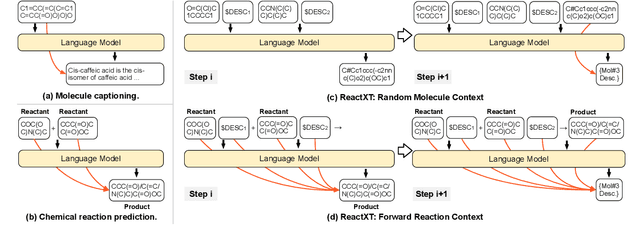

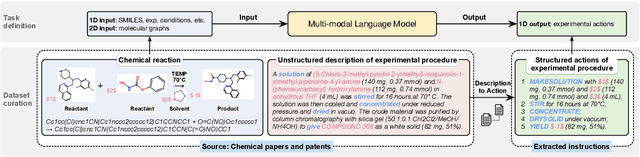
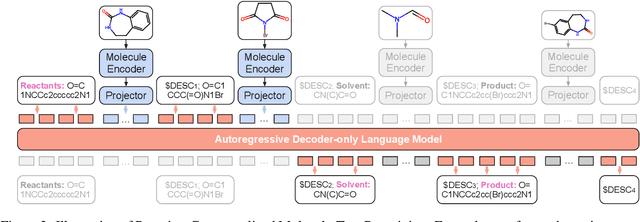
Molecule-text modeling, which aims to facilitate molecule-relevant tasks with a textual interface and textual knowledge, is an emerging research direction. Beyond single molecules, studying reaction-text modeling holds promise for helping the synthesis of new materials and drugs. However, previous works mostly neglect reaction-text modeling: they primarily focus on modeling individual molecule-text pairs or learning chemical reactions without texts in context. Additionally, one key task of reaction-text modeling -- experimental procedure prediction -- is less explored due to the absence of an open-source dataset. The task is to predict step-by-step actions of conducting chemical experiments and is crucial to automating chemical synthesis. To resolve the challenges above, we propose a new pretraining method, ReactXT, for reaction-text modeling, and a new dataset, OpenExp, for experimental procedure prediction. Specifically, ReactXT features three types of input contexts to incrementally pretrain LMs. Each of the three input contexts corresponds to a pretraining task to improve the text-based understanding of either reactions or single molecules. ReactXT demonstrates consistent improvements in experimental procedure prediction and molecule captioning and offers competitive results in retrosynthesis. Our code is available at https://github.com/syr-cn/ReactXT.
Rethinking Tokenizer and Decoder in Masked Graph Modeling for Molecules
Oct 23, 2023



Masked graph modeling excels in the self-supervised representation learning of molecular graphs. Scrutinizing previous studies, we can reveal a common scheme consisting of three key components: (1) graph tokenizer, which breaks a molecular graph into smaller fragments (i.e., subgraphs) and converts them into tokens; (2) graph masking, which corrupts the graph with masks; (3) graph autoencoder, which first applies an encoder on the masked graph to generate the representations, and then employs a decoder on the representations to recover the tokens of the original graph. However, the previous MGM studies focus extensively on graph masking and encoder, while there is limited understanding of tokenizer and decoder. To bridge the gap, we first summarize popular molecule tokenizers at the granularity of node, edge, motif, and Graph Neural Networks (GNNs), and then examine their roles as the MGM's reconstruction targets. Further, we explore the potential of adopting an expressive decoder in MGM. Our results show that a subgraph-level tokenizer and a sufficiently expressive decoder with remask decoding have a large impact on the encoder's representation learning. Finally, we propose a novel MGM method SimSGT, featuring a Simple GNN-based Tokenizer (SGT) and an effective decoding strategy. We empirically validate that our method outperforms the existing molecule self-supervised learning methods. Our codes and checkpoints are available at https://github.com/syr-cn/SimSGT.
ReLM: Leveraging Language Models for Enhanced Chemical Reaction Prediction
Oct 20, 2023



Predicting chemical reactions, a fundamental challenge in chemistry, involves forecasting the resulting products from a given reaction process. Conventional techniques, notably those employing Graph Neural Networks (GNNs), are often limited by insufficient training data and their inability to utilize textual information, undermining their applicability in real-world applications. In this work, we propose ReLM, a novel framework that leverages the chemical knowledge encoded in language models (LMs) to assist GNNs, thereby enhancing the accuracy of real-world chemical reaction predictions. To further enhance the model's robustness and interpretability, we incorporate the confidence score strategy, enabling the LMs to self-assess the reliability of their predictions. Our experimental results demonstrate that ReLM improves the performance of state-of-the-art GNN-based methods across various chemical reaction datasets, especially in out-of-distribution settings. Codes are available at https://github.com/syr-cn/ReLM.