 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAblateCell: A Reproduce-then-Ablate Agent for Virtual Cell Repositories
Apr 21, 2026Systematic ablations are essential to attribute performance gains in AI Virtual Cells, yet they are rarely performed because biological repositories are under-standardized and tightly coupled to domain-specific data and formats. While recent coding agents can translate ideas into implementations, they typically stop at producing code and lack a verifier that can reproduce strong baselines and rigorously test which components truly matter. We introduce AblateCell, a reproduce-then-ablate agent for virtual cell repositories that closes this verification gap. AblateCell first reproduces reported baselines end-to-end by auto-configuring environments, resolving dependency and data issues, and rerunning official evaluations while emitting verifiable artifacts. It then conducts closed-loop ablation by generating a graph of isolated repository mutations and adaptively selecting experiments under a reward that trades off performance impact and execution cost. Evaluated on three single-cell perturbation prediction repositories (CPA, GEARS, BioLORD), AblateCell achieves 88.9% (+29.9% to human expert) end-to-end workflow success and 93.3% (+53.3% to heuristic) accuracy in recovering ground-truth critical components. These results enable scalable, repository-grounded verification and attribution directly on biological codebases.
SCALE:Scalable Conditional Atlas-Level Endpoint transport for virtual cell perturbation prediction
Mar 19, 2026Virtual cell models aim to enable in silico experimentation by predicting how cells respond to genetic, chemical, or cytokine perturbations from single-cell measurements. In practice, however, large-scale perturbation prediction remains constrained by three coupled bottlenecks: inefficient training and inference pipelines, unstable modeling in high-dimensional sparse expression space, and evaluation protocols that overemphasize reconstruction-like accuracy while underestimating biological fidelity. In this work we present a specialized large-scale foundation model SCALE for virtual cell perturbation prediction that addresses the above limitations jointly. First, we build a BioNeMo-based training and inference framework that substantially improves data throughput, distributed scalability, and deployment efficiency, yielding 12.51* speedup on pretrain and 1.29* on inference over the prior SOTA pipeline under matched system settings. Second, we formulate perturbation prediction as conditional transport and implement it with a set-aware flow architecture that couples LLaMA-based cellular encoding with endpoint-oriented supervision. This design yields more stable training and stronger recovery of perturbation effects. Third, we evaluate the model on Tahoe-100M using a rigorous cell-level protocol centered on biologically meaningful metrics rather than reconstruction alone. On this benchmark, our model improves PDCorr by 12.02% and DE Overlap by 10.66% over STATE. Together, these results suggest that advancing virtual cells requires not only better generative objectives, but also the co-design of scalable infrastructure, stable transport modeling, and biologically faithful evaluation.
HarmonyCell: Automating Single-Cell Perturbation Modeling under Semantic and Distribution Shifts
Mar 02, 2026Single-cell perturbation studies face dual heterogeneity bottlenecks: (i) semantic heterogeneity--identical biological concepts encoded under incompatible metadata schemas across datasets; and (ii) statistical heterogeneity--distribution shifts from biological variation demanding dataset-specific inductive biases. We propose HarmonyCell, an end-to-end agent framework resolving each challenge through a dedicated mechanism: an LLM-driven Semantic Unifier autonomously maps disparate metadata into a canonical interface without manual intervention; and an adaptive Monte Carlo Tree Search engine operates over a hierarchical action space to synthesize architectures with optimal statistical inductive biases for distribution shifts. Evaluated across diverse perturbation tasks under both semantic and distribution shifts, HarmonyCell achieves a 95% valid execution rate on heterogeneous input datasets (versus 0% for general agents) while matching or even exceeding expert-designed baselines in rigorous out-of-distribution evaluations. This dual-track orchestration enables scalable automatic virtual cell modeling without dataset-specific engineering.
Lost in Tokenization: Context as the Key to Unlocking Biomolecular Understanding in Scientific LLMs
Oct 27, 2025Scientific Large Language Models (Sci-LLMs) have emerged as a promising frontier for accelerating biological discovery. However, these models face a fundamental challenge when processing raw biomolecular sequences: the tokenization dilemma. Whether treating sequences as a specialized language, risking the loss of functional motif information, or as a separate modality, introducing formidable alignment challenges, current strategies fundamentally limit their reasoning capacity. We challenge this sequence-centric paradigm by positing that a more effective strategy is to provide Sci-LLMs with high-level structured context derived from established bioinformatics tools, thereby bypassing the need to interpret low-level noisy sequence data directly. Through a systematic comparison of leading Sci-LLMs on biological reasoning tasks, we tested three input modes: sequence-only, context-only, and a combination of both. Our findings are striking: the context-only approach consistently and substantially outperforms all other modes. Even more revealing, the inclusion of the raw sequence alongside its high-level context consistently degrades performance, indicating that raw sequences act as informational noise, even for models with specialized tokenization schemes. These results suggest that the primary strength of existing Sci-LLMs lies not in their nascent ability to interpret biomolecular syntax from scratch, but in their profound capacity for reasoning over structured, human-readable knowledge. Therefore, we argue for reframing Sci-LLMs not as sequence decoders, but as powerful reasoning engines over expert knowledge. This work lays the foundation for a new class of hybrid scientific AI agents, repositioning the developmental focus from direct sequence interpretation towards high-level knowledge synthesis. The code is available at github.com/opendatalab-raise-dev/CoKE.
Protein-SE(3): Benchmarking SE(3)-based Generative Models for Protein Structure Design
Jul 27, 2025SE(3)-based generative models have shown great promise in protein geometry modeling and effective structure design. However, the field currently lacks a modularized benchmark to enable comprehensive investigation and fair comparison of different methods. In this paper, we propose Protein-SE(3), a new benchmark based on a unified training framework, which comprises protein scaffolding tasks, integrated generative models, high-level mathematical abstraction, and diverse evaluation metrics. Recent advanced generative models designed for protein scaffolding, from multiple perspectives like DDPM (Genie1 and Genie2), Score Matching (FrameDiff and RfDiffusion) and Flow Matching (FoldFlow and FrameFlow) are integrated into our framework. All integrated methods are fairly investigated with the same training dataset and evaluation metrics. Furthermore, we provide a high-level abstraction of the mathematical foundations behind the generative models, enabling fast prototyping of future algorithms without reliance on explicit protein structures. Accordingly, we release the first comprehensive benchmark built upon unified training framework for SE(3)-based protein structure design, which is publicly accessible at https://github.com/BruthYU/protein-se3.
AlphaFold Database Debiasing for Robust Inverse Folding
Jun 10, 2025The AlphaFold Protein Structure Database (AFDB) offers unparalleled structural coverage at near-experimental accuracy, positioning it as a valuable resource for data-driven protein design. However, its direct use in training deep models that are sensitive to fine-grained atomic geometry, such as inverse folding, exposes a critical limitation. Comparative analysis of structural feature distributions reveals that AFDB structures exhibit distinct statistical regularities, reflecting a systematic geometric bias that deviates from the conformational diversity found in experimentally determined structures from the Protein Data Bank (PDB). While AFDB structures are cleaner and more idealized, PDB structures capture the intrinsic variability and physical realism essential for generalization in downstream tasks. To address this discrepancy, we introduce a Debiasing Structure AutoEncoder (DeSAE) that learns to reconstruct native-like conformations from intentionally corrupted backbone geometries. By training the model to recover plausible structural states, DeSAE implicitly captures a more robust and natural structural manifold. At inference, applying DeSAE to AFDB structures produces debiased structures that significantly improve inverse folding performance across multiple benchmarks. This work highlights the critical impact of subtle systematic biases in predicted structures and presents a principled framework for debiasing, significantly boosting the performance of structure-based learning tasks like inverse folding.
G2PDiffusion: Genotype-to-Phenotype Prediction with Diffusion Models
Feb 07, 2025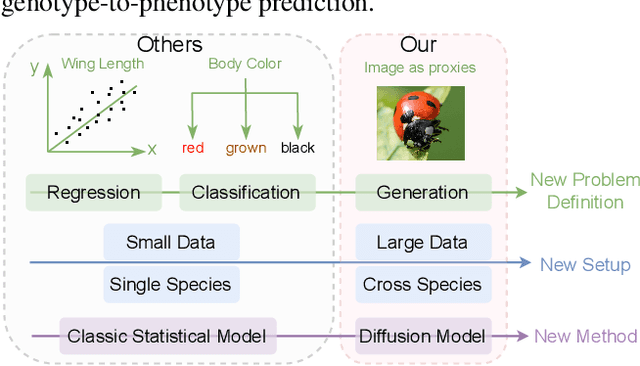
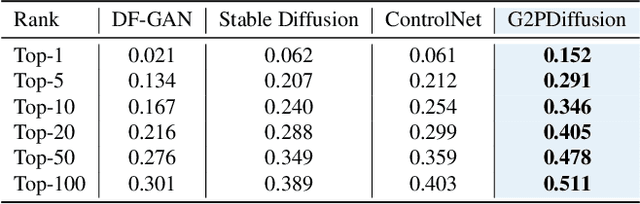
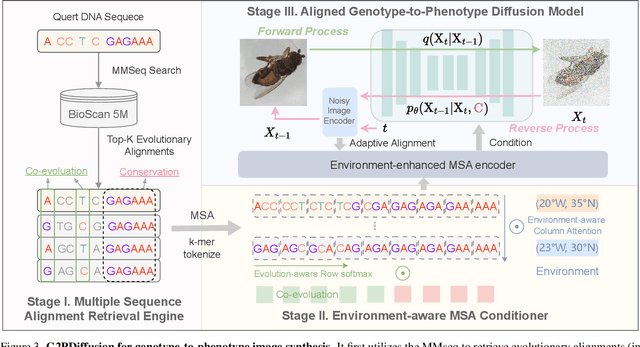
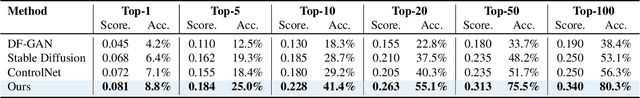
Discovering the genotype-phenotype relationship is crucial for genetic engineering, which will facilitate advances in fields such as crop breeding, conservation biology, and personalized medicine. Current research usually focuses on single species and small datasets due to limitations in phenotypic data collection, especially for traits that require visual assessments or physical measurements. Deciphering complex and composite phenotypes, such as morphology, from genetic data at scale remains an open question. To break through traditional generic models that rely on simplified assumptions, this paper introduces G2PDiffusion, the first-of-its-kind diffusion model designed for genotype-to-phenotype generation across multiple species. Specifically, we use images to represent morphological phenotypes across species and redefine phenotype prediction as conditional image generation. To this end, this paper introduces an environment-enhanced DNA sequence conditioner and trains a stable diffusion model with a novel alignment method to improve genotype-to-phenotype consistency. Extensive experiments demonstrate that our approach enhances phenotype prediction accuracy across species, capturing subtle genetic variations that contribute to observable traits.
MeToken: Uniform Micro-environment Token Boosts Post-Translational Modification Prediction
Nov 04, 2024



Post-translational modifications (PTMs) profoundly expand the complexity and functionality of the proteome, regulating protein attributes and interactions that are crucial for biological processes. Accurately predicting PTM sites and their specific types is therefore essential for elucidating protein function and understanding disease mechanisms. Existing computational approaches predominantly focus on protein sequences to predict PTM sites, driven by the recognition of sequence-dependent motifs. However, these approaches often overlook protein structural contexts. In this work, we first compile a large-scale sequence-structure PTM dataset, which serves as the foundation for fair comparison. We introduce the MeToken model, which tokenizes the micro-environment of each amino acid, integrating both sequence and structural information into unified discrete tokens. This model not only captures the typical sequence motifs associated with PTMs but also leverages the spatial arrangements dictated by protein tertiary structures, thus providing a holistic view of the factors influencing PTM sites. Designed to address the long-tail distribution of PTM types, MeToken employs uniform sub-codebooks that ensure even the rarest PTMs are adequately represented and distinguished. We validate the effectiveness and generalizability of MeToken across multiple datasets, demonstrating its superior performance in accurately identifying PTM types. The results underscore the importance of incorporating structural data and highlight MeToken's potential in facilitating accurate and comprehensive PTM predictions, which could significantly impact proteomics research. The code and datasets are available at https://github.com/A4Bio/MeToken.
FlexMol: A Flexible Toolkit for Benchmarking Molecular Relational Learning
Oct 19, 2024



Molecular relational learning (MRL) is crucial for understanding the interaction behaviors between molecular pairs, a critical aspect of drug discovery and development. However, the large feasible model space of MRL poses significant challenges to benchmarking, and existing MRL frameworks face limitations in flexibility and scope. To address these challenges, avoid repetitive coding efforts, and ensure fair comparison of models, we introduce FlexMol, a comprehensive toolkit designed to facilitate the construction and evaluation of diverse model architectures across various datasets and performance metrics. FlexMol offers a robust suite of preset model components, including 16 drug encoders, 13 protein sequence encoders, 9 protein structure encoders, and 7 interaction layers. With its easy-to-use API and flexibility, FlexMol supports the dynamic construction of over 70, 000 distinct combinations of model architectures. Additionally, we provide detailed benchmark results and code examples to demonstrate FlexMol's effectiveness in simplifying and standardizing MRL model development and comparison.
Peer Review as A Multi-Turn and Long-Context Dialogue with Role-Based Interactions
Jun 09, 2024Large Language Models (LLMs) have demonstrated wide-ranging applications across various fields and have shown significant potential in the academic peer-review process. However, existing applications are primarily limited to static review generation based on submitted papers, which fail to capture the dynamic and iterative nature of real-world peer reviews. In this paper, we reformulate the peer-review process as a multi-turn, long-context dialogue, incorporating distinct roles for authors, reviewers, and decision makers. We construct a comprehensive dataset containing over 26,841 papers with 92,017 reviews collected from multiple sources, including the top-tier conference and prestigious journal. This dataset is meticulously designed to facilitate the applications of LLMs for multi-turn dialogues, effectively simulating the complete peer-review process. Furthermore, we propose a series of metrics to evaluate the performance of LLMs for each role under this reformulated peer-review setting, ensuring fair and comprehensive evaluations. We believe this work provides a promising perspective on enhancing the LLM-driven peer-review process by incorporating dynamic, role-based interactions. It aligns closely with the iterative and interactive nature of real-world academic peer review, offering a robust foundation for future research and development in this area. We open-source the dataset at https://github.com/chengtan9907/ReviewMT.