 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeUniIF: Unified Molecule Inverse Folding
May 29, 2024
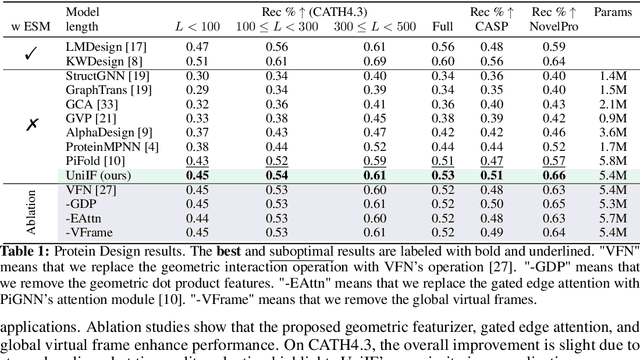
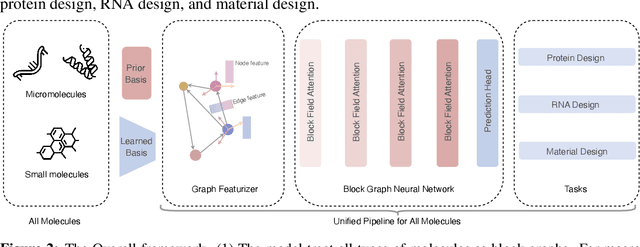

Molecule inverse folding has been a long-standing challenge in chemistry and biology, with the potential to revolutionize drug discovery and material science. Despite specified models have been proposed for different small- or macro-molecules, few have attempted to unify the learning process, resulting in redundant efforts. Complementary to recent advancements in molecular structure prediction, such as RoseTTAFold All-Atom and AlphaFold3, we propose the unified model UniIF for the inverse folding of all molecules. We do such unification in two levels: 1) Data-Level: We propose a unified block graph data form for all molecules, including the local frame building and geometric feature initialization. 2) Model-Level: We introduce a geometric block attention network, comprising a geometric interaction, interactive attention and virtual long-term dependency modules, to capture the 3D interactions of all molecules. Through comprehensive evaluations across various tasks such as protein design, RNA design, and material design, we demonstrate that our proposed method surpasses state-of-the-art methods on all tasks. UniIF offers a versatile and effective solution for general molecule inverse folding.