 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeDual Latent Memory for Visual Multi-agent System
Jan 31, 2026While Visual Multi-Agent Systems (VMAS) promise to enhance comprehensive abilities through inter-agent collaboration, empirical evidence reveals a counter-intuitive "scaling wall": increasing agent turns often degrades performance while exponentially inflating token costs. We attribute this failure to the information bottleneck inherent in text-centric communication, where converting perceptual and thinking trajectories into discrete natural language inevitably induces semantic loss. To this end, we propose L$^{2}$-VMAS, a novel model-agnostic framework that enables inter-agent collaboration with dual latent memories. Furthermore, we decouple the perception and thinking while dynamically synthesizing dual latent memories. Additionally, we introduce an entropy-driven proactive triggering that replaces passive information transmission with efficient, on-demand memory access. Extensive experiments among backbones, sizes, and multi-agent structures demonstrate that our method effectively breaks the "scaling wall" with superb scalability, improving average accuracy by 2.7-5.4% while reducing token usage by 21.3-44.8%. Codes: https://github.com/YU-deep/L2-VMAS.
Chain-of-Thought Compression Should Not Be Blind: V-Skip for Efficient Multimodal Reasoning via Dual-Path Anchoring
Jan 21, 2026While Chain-of-Thought (CoT) reasoning significantly enhances the performance of Multimodal Large Language Models (MLLMs), its autoregressive nature incurs prohibitive latency constraints. Current efforts to mitigate this via token compression often fail by blindly applying text-centric metrics to multimodal contexts. We identify a critical failure mode termed Visual Amnesia, where linguistically redundant tokens are erroneously pruned, leading to hallucinations. To address this, we introduce V-Skip that reformulates token pruning as a Visual-Anchored Information Bottleneck (VA-IB) optimization problem. V-Skip employs a dual-path gating mechanism that weighs token importance through both linguistic surprisal and cross-modal attention flow, effectively rescuing visually salient anchors. Extensive experiments on Qwen2-VL and Llama-3.2 families demonstrate that V-Skip achieves a $2.9\times$ speedup with negligible accuracy loss. Specifically, it preserves fine-grained visual details, outperforming other baselines over 30\% on the DocVQA.
SCP: Accelerating Discovery with a Global Web of Autonomous Scientific Agents
Dec 30, 2025We introduce SCP: the Science Context Protocol, an open-source standard designed to accelerate discovery by enabling a global network of autonomous scientific agents. SCP is built on two foundational pillars: (1) Unified Resource Integration: At its core, SCP provides a universal specification for describing and invoking scientific resources, spanning software tools, models, datasets, and physical instruments. This protocol-level standardization enables AI agents and applications to discover, call, and compose capabilities seamlessly across disparate platforms and institutional boundaries. (2) Orchestrated Experiment Lifecycle Management: SCP complements the protocol with a secure service architecture, which comprises a centralized SCP Hub and federated SCP Servers. This architecture manages the complete experiment lifecycle (registration, planning, execution, monitoring, and archival), enforces fine-grained authentication and authorization, and orchestrates traceable, end-to-end workflows that bridge computational and physical laboratories. Based on SCP, we have constructed a scientific discovery platform that offers researchers and agents a large-scale ecosystem of more than 1,600 tool resources. Across diverse use cases, SCP facilitates secure, large-scale collaboration between heterogeneous AI systems and human researchers while significantly reducing integration overhead and enhancing reproducibility. By standardizing scientific context and tool orchestration at the protocol level, SCP establishes essential infrastructure for scalable, multi-institution, agent-driven science.
Socrates-Mol: Self-Oriented Cognitive Reasoning through Autonomous Trial-and-Error with Empirical-Bayesian Screening for Molecules
Nov 14, 2025Molecular property prediction is fundamental to chemical engineering applications such as solvent screening. We present Socrates-Mol, a framework that transforms language models into empirical Bayesian reasoners through context engineering, addressing cold start problems without model fine-tuning. The system implements a reflective-prediction cycle where initial outputs serve as priors, retrieved molecular cases provide evidence, and refined predictions form posteriors, extracting reusable chemical rules from sparse data. We introduce ranking tasks aligned with industrial screening priorities and employ cross-model self-consistency across five language models to reduce variance. Experiments on amine solvent LogP prediction reveal task-dependent patterns: regression achieves 72% MAE reduction and 112% R-squared improvement through self-consistency, while ranking tasks show limited gains due to systematic multi-model biases. The framework reduces deployment costs by over 70% compared to full fine-tuning, providing a scalable solution for molecular property prediction while elucidating the task-adaptive nature of self-consistency mechanisms.
GGBench: A Geometric Generative Reasoning Benchmark for Unified Multimodal Models
Nov 14, 2025
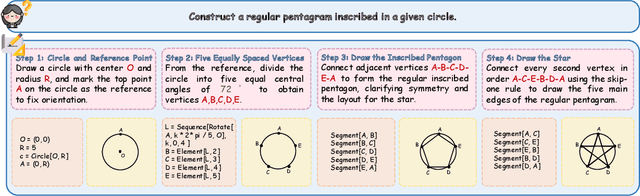

The advent of Unified Multimodal Models (UMMs) signals a paradigm shift in artificial intelligence, moving from passive perception to active, cross-modal generation. Despite their unprecedented ability to synthesize information, a critical gap persists in evaluation: existing benchmarks primarily assess discriminative understanding or unconstrained image generation separately, failing to measure the integrated cognitive process of generative reasoning. To bridge this gap, we propose that geometric construction provides an ideal testbed as it inherently demands a fusion of language comprehension and precise visual generation. We introduce GGBench, a benchmark designed specifically to evaluate geometric generative reasoning. It provides a comprehensive framework for systematically diagnosing a model's ability to not only understand and reason but to actively construct a solution, thereby setting a more rigorous standard for the next generation of intelligent systems. Project website: https://opendatalab-raiser.github.io/GGBench/.
Lost in Tokenization: Context as the Key to Unlocking Biomolecular Understanding in Scientific LLMs
Oct 27, 2025Scientific Large Language Models (Sci-LLMs) have emerged as a promising frontier for accelerating biological discovery. However, these models face a fundamental challenge when processing raw biomolecular sequences: the tokenization dilemma. Whether treating sequences as a specialized language, risking the loss of functional motif information, or as a separate modality, introducing formidable alignment challenges, current strategies fundamentally limit their reasoning capacity. We challenge this sequence-centric paradigm by positing that a more effective strategy is to provide Sci-LLMs with high-level structured context derived from established bioinformatics tools, thereby bypassing the need to interpret low-level noisy sequence data directly. Through a systematic comparison of leading Sci-LLMs on biological reasoning tasks, we tested three input modes: sequence-only, context-only, and a combination of both. Our findings are striking: the context-only approach consistently and substantially outperforms all other modes. Even more revealing, the inclusion of the raw sequence alongside its high-level context consistently degrades performance, indicating that raw sequences act as informational noise, even for models with specialized tokenization schemes. These results suggest that the primary strength of existing Sci-LLMs lies not in their nascent ability to interpret biomolecular syntax from scratch, but in their profound capacity for reasoning over structured, human-readable knowledge. Therefore, we argue for reframing Sci-LLMs not as sequence decoders, but as powerful reasoning engines over expert knowledge. This work lays the foundation for a new class of hybrid scientific AI agents, repositioning the developmental focus from direct sequence interpretation towards high-level knowledge synthesis. The code is available at github.com/opendatalab-raise-dev/CoKE.
ResearchPulse: Building Method-Experiment Chains through Multi-Document Scientific Inference
Sep 03, 2025Understanding how scientific ideas evolve requires more than summarizing individual papers-it demands structured, cross-document reasoning over thematically related research. In this work, we formalize multi-document scientific inference, a new task that extracts and aligns motivation, methodology, and experimental results across related papers to reconstruct research development chains. This task introduces key challenges, including temporally aligning loosely structured methods and standardizing heterogeneous experimental tables. We present ResearchPulse, an agent-based framework that integrates instruction planning, scientific content extraction, and structured visualization. It consists of three coordinated agents: a Plan Agent for task decomposition, a Mmap-Agent that constructs motivation-method mind maps, and a Lchart-Agent that synthesizes experimental line charts. To support this task, we introduce ResearchPulse-Bench, a citation-aware benchmark of annotated paper clusters. Experiments show that our system, despite using 7B-scale agents, consistently outperforms strong baselines like GPT-4o in semantic alignment, structural consistency, and visual fidelity. The dataset are available in https://huggingface.co/datasets/ResearchPulse/ResearchPulse-Bench.
A Survey of Scientific Large Language Models: From Data Foundations to Agent Frontiers
Aug 28, 2025



Scientific Large Language Models (Sci-LLMs) are transforming how knowledge is represented, integrated, and applied in scientific research, yet their progress is shaped by the complex nature of scientific data. This survey presents a comprehensive, data-centric synthesis that reframes the development of Sci-LLMs as a co-evolution between models and their underlying data substrate. We formulate a unified taxonomy of scientific data and a hierarchical model of scientific knowledge, emphasizing the multimodal, cross-scale, and domain-specific challenges that differentiate scientific corpora from general natural language processing datasets. We systematically review recent Sci-LLMs, from general-purpose foundations to specialized models across diverse scientific disciplines, alongside an extensive analysis of over 270 pre-/post-training datasets, showing why Sci-LLMs pose distinct demands -- heterogeneous, multi-scale, uncertainty-laden corpora that require representations preserving domain invariance and enabling cross-modal reasoning. On evaluation, we examine over 190 benchmark datasets and trace a shift from static exams toward process- and discovery-oriented assessments with advanced evaluation protocols. These data-centric analyses highlight persistent issues in scientific data development and discuss emerging solutions involving semi-automated annotation pipelines and expert validation. Finally, we outline a paradigm shift toward closed-loop systems where autonomous agents based on Sci-LLMs actively experiment, validate, and contribute to a living, evolving knowledge base. Collectively, this work provides a roadmap for building trustworthy, continually evolving artificial intelligence (AI) systems that function as a true partner in accelerating scientific discovery.
StructVRM: Aligning Multimodal Reasoning with Structured and Verifiable Reward Models
Aug 07, 2025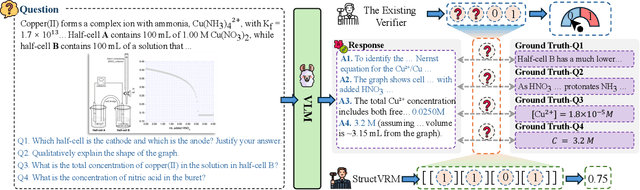

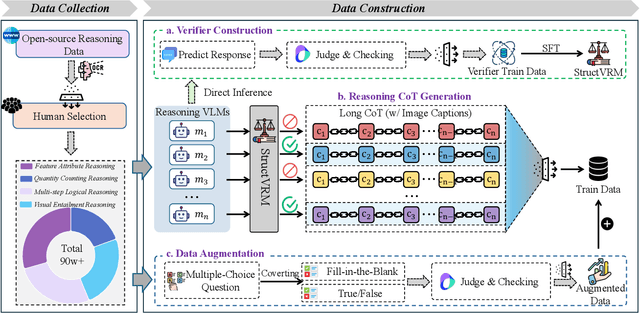
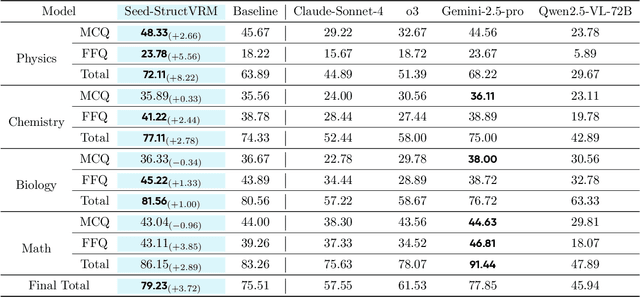
Existing Vision-Language Models often struggle with complex, multi-question reasoning tasks where partial correctness is crucial for effective learning. Traditional reward mechanisms, which provide a single binary score for an entire response, are too coarse to guide models through intricate problems with multiple sub-parts. To address this, we introduce StructVRM, a method that aligns multimodal reasoning with Structured and Verifiable Reward Models. At its core is a model-based verifier trained to provide fine-grained, sub-question-level feedback, assessing semantic and mathematical equivalence rather than relying on rigid string matching. This allows for nuanced, partial credit scoring in previously intractable problem formats. Extensive experiments demonstrate the effectiveness of StructVRM. Our trained model, Seed-StructVRM, achieves state-of-the-art performance on six out of twelve public multimodal benchmarks and our newly curated, high-difficulty STEM-Bench. The success of StructVRM validates that training with structured, verifiable rewards is a highly effective approach for advancing the capabilities of multimodal models in complex, real-world reasoning domains.
Magentic-UI: Towards Human-in-the-loop Agentic Systems
Jul 30, 2025AI agents powered by large language models are increasingly capable of autonomously completing complex, multi-step tasks using external tools. Yet, they still fall short of human-level performance in most domains including computer use, software development, and research. Their growing autonomy and ability to interact with the outside world, also introduces safety and security risks including potentially misaligned actions and adversarial manipulation. We argue that human-in-the-loop agentic systems offer a promising path forward, combining human oversight and control with AI efficiency to unlock productivity from imperfect systems. We introduce Magentic-UI, an open-source web interface for developing and studying human-agent interaction. Built on a flexible multi-agent architecture, Magentic-UI supports web browsing, code execution, and file manipulation, and can be extended with diverse tools via Model Context Protocol (MCP). Moreover, Magentic-UI presents six interaction mechanisms for enabling effective, low-cost human involvement: co-planning, co-tasking, multi-tasking, action guards, and long-term memory. We evaluate Magentic-UI across four dimensions: autonomous task completion on agentic benchmarks, simulated user testing of its interaction capabilities, qualitative studies with real users, and targeted safety assessments. Our findings highlight Magentic-UI's potential to advance safe and efficient human-agent collaboration.