 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Networks in Modern AI-aided Drug Discovery
Jun 07, 2025Graph neural networks (GNNs), as topology/structure-aware models within deep learning, have emerged as powerful tools for AI-aided drug discovery (AIDD). By directly operating on molecular graphs, GNNs offer an intuitive and expressive framework for learning the complex topological and geometric features of drug-like molecules, cementing their role in modern molecular modeling. This review provides a comprehensive overview of the methodological foundations and representative applications of GNNs in drug discovery, spanning tasks such as molecular property prediction, virtual screening, molecular generation, biomedical knowledge graph construction, and synthesis planning. Particular attention is given to recent methodological advances, including geometric GNNs, interpretable models, uncertainty quantification, scalable graph architectures, and graph generative frameworks. We also discuss how these models integrate with modern deep learning approaches, such as self-supervised learning, multi-task learning, meta-learning and pre-training. Throughout this review, we highlight the practical challenges and methodological bottlenecks encountered when applying GNNs to real-world drug discovery pipelines, and conclude with a discussion on future directions.
Tokenizing Electron Cloud in Protein-Ligand Interaction Learning
May 25, 2025



The affinity and specificity of protein-molecule binding directly impact functional outcomes, uncovering the mechanisms underlying biological regulation and signal transduction. Most deep-learning-based prediction approaches focus on structures of atoms or fragments. However, quantum chemical properties, such as electronic structures, are the key to unveiling interaction patterns but remain largely underexplored. To bridge this gap, we propose ECBind, a method for tokenizing electron cloud signals into quantized embeddings, enabling their integration into downstream tasks such as binding affinity prediction. By incorporating electron densities, ECBind helps uncover binding modes that cannot be fully represented by atom-level models. Specifically, to remove the redundancy inherent in electron cloud signals, a structure-aware transformer and hierarchical codebooks encode 3D binding sites enriched with electron structures into tokens. These tokenized codes are then used for specific tasks with labels. To extend its applicability to a wider range of scenarios, we utilize knowledge distillation to develop an electron-cloud-agnostic prediction model. Experimentally, ECBind demonstrates state-of-the-art performance across multiple tasks, achieving improvements of 6.42\% and 15.58\% in per-structure Pearson and Spearman correlation coefficients, respectively.
Reaction-conditioned De Novo Enzyme Design with GENzyme
Nov 10, 2024The introduction of models like RFDiffusionAA, AlphaFold3, AlphaProteo, and Chai1 has revolutionized protein structure modeling and interaction prediction, primarily from a binding perspective, focusing on creating ideal lock-and-key models. However, these methods can fall short for enzyme-substrate interactions, where perfect binding models are rare, and induced fit states are more common. To address this, we shift to a functional perspective for enzyme design, where the enzyme function is defined by the reaction it catalyzes. Here, we introduce \textsc{GENzyme}, a \textit{de novo} enzyme design model that takes a catalytic reaction as input and generates the catalytic pocket, full enzyme structure, and enzyme-substrate binding complex. \textsc{GENzyme} is an end-to-end, three-staged model that integrates (1) a catalytic pocket generation and sequence co-design module, (2) a pocket inpainting and enzyme inverse folding module, and (3) a binding and screening module to optimize and predict enzyme-substrate complexes. The entire design process is driven by the catalytic reaction being targeted. This reaction-first approach allows for more accurate and biologically relevant enzyme design, potentially surpassing structure-based and binding-focused models in creating enzymes capable of catalyzing specific reactions. We provide \textsc{GENzyme} code at https://github.com/WillHua127/GENzyme.
Token-Mol 1.0: Tokenized drug design with large language model
Jul 10, 2024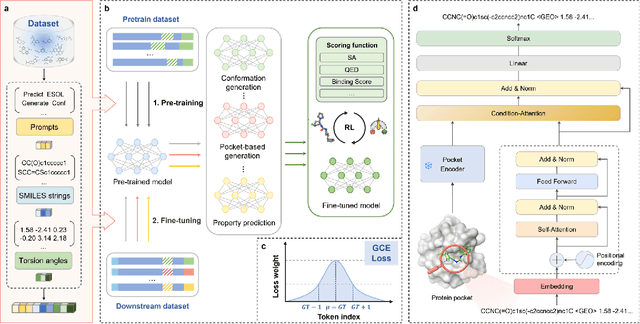

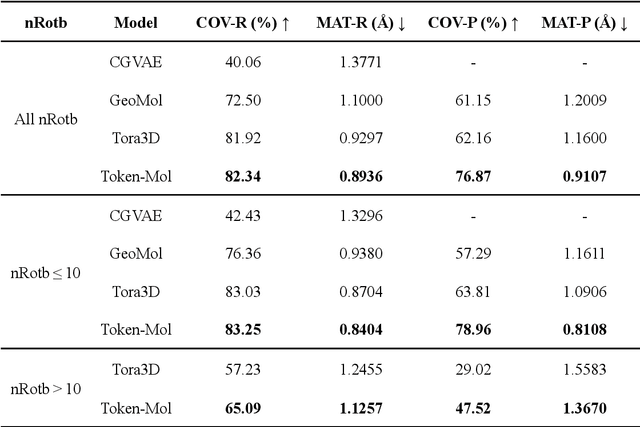
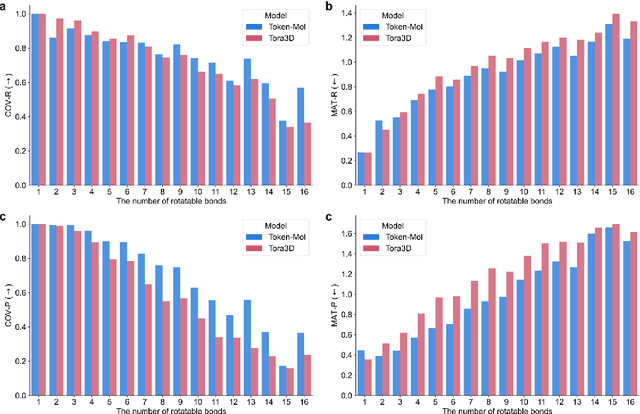
Significant interests have recently risen in leveraging sequence-based large language models (LLMs) for drug design. However, most current applications of LLMs in drug discovery lack the ability to comprehend three-dimensional (3D) structures, thereby limiting their effectiveness in tasks that explicitly involve molecular conformations. In this study, we introduced Token-Mol, a token-only 3D drug design model. This model encodes all molecular information, including 2D and 3D structures, as well as molecular property data, into tokens, which transforms classification and regression tasks in drug discovery into probabilistic prediction problems, thereby enabling learning through a unified paradigm. Token-Mol is built on the transformer decoder architecture and trained using random causal masking techniques. Additionally, we proposed the Gaussian cross-entropy (GCE) loss function to overcome the challenges in regression tasks, significantly enhancing the capacity of LLMs to learn continuous numerical values. Through a combination of fine-tuning and reinforcement learning (RL), Token-Mol achieves performance comparable to or surpassing existing task-specific methods across various downstream tasks, including pocket-based molecular generation, conformation generation, and molecular property prediction. Compared to existing molecular pre-trained models, Token-Mol exhibits superior proficiency in handling a wider range of downstream tasks essential for drug design. Notably, our approach improves regression task accuracy by approximately 30% compared to similar token-only methods. Token-Mol overcomes the precision limitations of token-only models and has the potential to integrate seamlessly with general models such as ChatGPT, paving the way for the development of a universal artificial intelligence drug design model that facilitates rapid and high-quality drug design by experts.
Rethinking the Diffusion Models for Numerical Tabular Data Imputation from the Perspective of Wasserstein Gradient Flow
Jun 22, 2024



Diffusion models (DMs) have gained attention in Missing Data Imputation (MDI), but there remain two long-neglected issues to be addressed: (1). Inaccurate Imputation, which arises from inherently sample-diversification-pursuing generative process of DMs. (2). Difficult Training, which stems from intricate design required for the mask matrix in model training stage. To address these concerns within the realm of numerical tabular datasets, we introduce a novel principled approach termed Kernelized Negative Entropy-regularized Wasserstein gradient flow Imputation (KnewImp). Specifically, based on Wasserstein gradient flow (WGF) framework, we first prove that issue (1) stems from the cost functionals implicitly maximized in DM-based MDI are equivalent to the MDI's objective plus diversification-promoting non-negative terms. Based on this, we then design a novel cost functional with diversification-discouraging negative entropy and derive our KnewImp approach within WGF framework and reproducing kernel Hilbert space. After that, we prove that the imputation procedure of KnewImp can be derived from another cost functional related to the joint distribution, eliminating the need for the mask matrix and hence naturally addressing issue (2). Extensive experiments demonstrate that our proposed KnewImp approach significantly outperforms existing state-of-the-art methods.
CBGBench: Fill in the Blank of Protein-Molecule Complex Binding Graph
Jun 16, 2024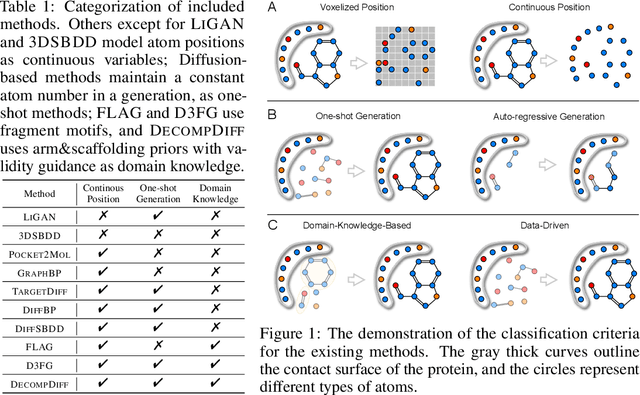



Structure-based drug design (SBDD) aims to generate potential drugs that can bind to a target protein and is greatly expedited by the aid of AI techniques in generative models. However, a lack of systematic understanding persists due to the diverse settings, complex implementation, difficult reproducibility, and task singularity. Firstly, the absence of standardization can lead to unfair comparisons and inconclusive insights. To address this dilemma, we propose CBGBench, a comprehensive benchmark for SBDD, that unifies the task as a generative heterogeneous graph completion, analogous to fill-in-the-blank of the 3D complex binding graph. By categorizing existing methods based on their attributes, CBGBench facilitates a modular and extensible framework that implements various cutting-edge methods. Secondly, a single task on \textit{de novo} molecule generation can hardly reflect their capabilities. To broaden the scope, we have adapted these models to a range of tasks essential in drug design, which are considered sub-tasks within the graph fill-in-the-blank tasks. These tasks include the generative designation of \textit{de novo} molecules, linkers, fragments, scaffolds, and sidechains, all conditioned on the structures of protein pockets. Our evaluations are conducted with fairness, encompassing comprehensive perspectives on interaction, chemical properties, geometry authenticity, and substructure validity. We further provide the pre-trained versions of the state-of-the-art models and deep insights with analysis from empirical studies. The codebase for CBGBench is publicly accessible at \url{https://github.com/Edapinenut/CBGBench}.
Deep Lead Optimization: Leveraging Generative AI for Structural Modification
Apr 30, 2024The idea of using deep-learning-based molecular generation to accelerate discovery of drug candidates has attracted extraordinary attention, and many deep generative models have been developed for automated drug design, termed molecular generation. In general, molecular generation encompasses two main strategies: de novo design, which generates novel molecular structures from scratch, and lead optimization, which refines existing molecules into drug candidates. Among them, lead optimization plays an important role in real-world drug design. For example, it can enable the development of me-better drugs that are chemically distinct yet more effective than the original drugs. It can also facilitate fragment-based drug design, transforming virtual-screened small ligands with low affinity into first-in-class medicines. Despite its importance, automated lead optimization remains underexplored compared to the well-established de novo generative models, due to its reliance on complex biological and chemical knowledge. To bridge this gap, we conduct a systematic review of traditional computational methods for lead optimization, organizing these strategies into four principal sub-tasks with defined inputs and outputs. This review delves into the basic concepts, goals, conventional CADD techniques, and recent advancements in AIDD. Additionally, we introduce a unified perspective based on constrained subgraph generation to harmonize the methodologies of de novo design and lead optimization. Through this lens, de novo design can incorporate strategies from lead optimization to address the challenge of generating hard-to-synthesize molecules; inversely, lead optimization can benefit from the innovations in de novo design by approaching it as a task of generating molecules conditioned on certain substructures.
Re-Dock: Towards Flexible and Realistic Molecular Docking with Diffusion Bridge
Feb 21, 2024Accurate prediction of protein-ligand binding structures, a task known as molecular docking is crucial for drug design but remains challenging. While deep learning has shown promise, existing methods often depend on holo-protein structures (docked, and not accessible in realistic tasks) or neglect pocket sidechain conformations, leading to limited practical utility and unrealistic conformation predictions. To fill these gaps, we introduce an under-explored task, named flexible docking to predict poses of ligand and pocket sidechains simultaneously and introduce Re-Dock, a novel diffusion bridge generative model extended to geometric manifolds. Specifically, we propose energy-to-geometry mapping inspired by the Newton-Euler equation to co-model the binding energy and conformations for reflecting the energy-constrained docking generative process. Comprehensive experiments on designed benchmark datasets including apo-dock and cross-dock demonstrate our model's superior effectiveness and efficiency over current methods.
Protein 3D Graph Structure Learning for Robust Structure-based Protein Property Prediction
Oct 19, 2023Protein structure-based property prediction has emerged as a promising approach for various biological tasks, such as protein function prediction and sub-cellular location estimation. The existing methods highly rely on experimental protein structure data and fail in scenarios where these data are unavailable. Predicted protein structures from AI tools (e.g., AlphaFold2) were utilized as alternatives. However, we observed that current practices, which simply employ accurately predicted structures during inference, suffer from notable degradation in prediction accuracy. While similar phenomena have been extensively studied in general fields (e.g., Computer Vision) as model robustness, their impact on protein property prediction remains unexplored. In this paper, we first investigate the reason behind the performance decrease when utilizing predicted structures, attributing it to the structure embedding bias from the perspective of structure representation learning. To study this problem, we identify a Protein 3D Graph Structure Learning Problem for Robust Protein Property Prediction (PGSL-RP3), collect benchmark datasets, and present a protein Structure embedding Alignment Optimization framework (SAO) to mitigate the problem of structure embedding bias between the predicted and experimental protein structures. Extensive experiments have shown that our framework is model-agnostic and effective in improving the property prediction of both predicted structures and experimental structures. The benchmark datasets and codes will be released to benefit the community.