 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeToward Closed-loop Molecular Discovery via Language Model, Property Alignment and Strategic Search
Dec 18, 2025Drug discovery is a time-consuming and expensive process, with traditional high-throughput and docking-based virtual screening hampered by low success rates and limited scalability. Recent advances in generative modelling, including autoregressive, diffusion, and flow-based approaches, have enabled de novo ligand design beyond the limits of enumerative screening. Yet these models often suffer from inadequate generalization, limited interpretability, and an overemphasis on binding affinity at the expense of key pharmacological properties, thereby restricting their translational utility. Here we present Trio, a molecular generation framework integrating fragment-based molecular language modeling, reinforcement learning, and Monte Carlo tree search, for effective and interpretable closed-loop targeted molecular design. Through the three key components, Trio enables context-aware fragment assembly, enforces physicochemical and synthetic feasibility, and guides a balanced search between the exploration of novel chemotypes and the exploitation of promising intermediates within protein binding pockets. Experimental results show that Trio reliably achieves chemically valid and pharmacologically enhanced ligands, outperforming state-of-the-art approaches with improved binding affinity (+7.85%), drug-likeness (+11.10%) and synthetic accessibility (+12.05%), while expanding molecular diversity more than fourfold. By combining generalization, plausibility, and interpretability, Trio establishes a closed-loop generative paradigm that redefines how chemical space can be navigated, offering a transformative foundation for the next era of AI-driven drug discovery.
Graph Neural Networks in Modern AI-aided Drug Discovery
Jun 07, 2025Graph neural networks (GNNs), as topology/structure-aware models within deep learning, have emerged as powerful tools for AI-aided drug discovery (AIDD). By directly operating on molecular graphs, GNNs offer an intuitive and expressive framework for learning the complex topological and geometric features of drug-like molecules, cementing their role in modern molecular modeling. This review provides a comprehensive overview of the methodological foundations and representative applications of GNNs in drug discovery, spanning tasks such as molecular property prediction, virtual screening, molecular generation, biomedical knowledge graph construction, and synthesis planning. Particular attention is given to recent methodological advances, including geometric GNNs, interpretable models, uncertainty quantification, scalable graph architectures, and graph generative frameworks. We also discuss how these models integrate with modern deep learning approaches, such as self-supervised learning, multi-task learning, meta-learning and pre-training. Throughout this review, we highlight the practical challenges and methodological bottlenecks encountered when applying GNNs to real-world drug discovery pipelines, and conclude with a discussion on future directions.
ProtFlow: Fast Protein Sequence Design via Flow Matching on Compressed Protein Language Model Embeddings
Apr 15, 2025The design of protein sequences with desired functionalities is a fundamental task in protein engineering. Deep generative methods, such as autoregressive models and diffusion models, have greatly accelerated the discovery of novel protein sequences. However, these methods mainly focus on local or shallow residual semantics and suffer from low inference efficiency, large modeling space and high training cost. To address these challenges, we introduce ProtFlow, a fast flow matching-based protein sequence design framework that operates on embeddings derived from semantically meaningful latent space of protein language models. By compressing and smoothing the latent space, ProtFlow enhances performance while training on limited computational resources. Leveraging reflow techniques, ProtFlow enables high-quality single-step sequence generation. Additionally, we develop a joint design pipeline for the design scene of multichain proteins. We evaluate ProtFlow across diverse protein design tasks, including general peptides and long-chain proteins, antimicrobial peptides, and antibodies. Experimental results demonstrate that ProtFlow outperforms task-specific methods in these applications, underscoring its potential and broad applicability in computational protein sequence design and analysis.
S$^2$ALM: Sequence-Structure Pre-trained Large Language Model for Comprehensive Antibody Representation Learning
Nov 20, 2024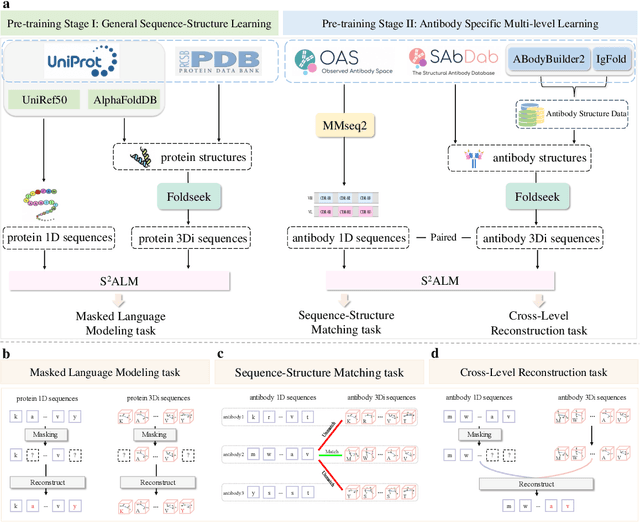
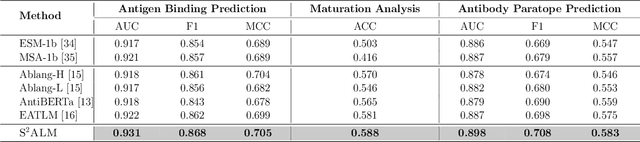
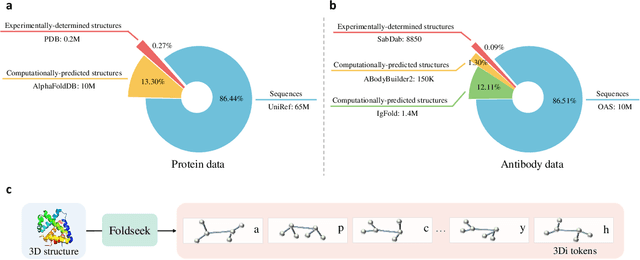
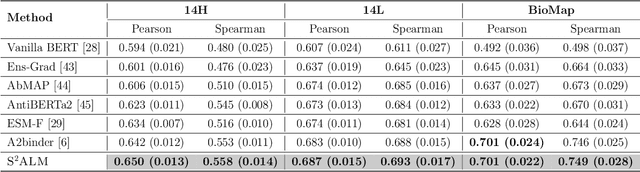
Antibodies safeguard our health through their precise and potent binding to specific antigens, demonstrating promising therapeutic efficacy in the treatment of numerous diseases, including COVID-19. Recent advancements in biomedical language models have shown the great potential to interpret complex biological structures and functions. However, existing antibody specific models have a notable limitation that they lack explicit consideration for antibody structural information, despite the fact that both 1D sequence and 3D structure carry unique and complementary insights into antibody behavior and functionality. This paper proposes Sequence-Structure multi-level pre-trained Antibody Language Model (S$^2$ALM), combining holistic sequential and structural information in one unified, generic antibody foundation model. We construct a hierarchical pre-training paradigm incorporated with two customized multi-level training objectives to facilitate the modeling of comprehensive antibody representations. S$^2$ALM's representation space uncovers inherent functional binding mechanisms, biological evolution properties and structural interaction patterns. Pre-trained over 75 million sequences and 11.7 million structures, S$^2$ALM can be adopted for diverse downstream tasks: accurately predicting antigen-antibody binding affinities, precisely distinguishing B cell maturation stages, identifying antibody crucial binding positions, and specifically designing novel coronavirus-binding antibodies. Remarkably, S$^2$ALM outperforms well-established and renowned baselines and sets new state-of-the-art performance across extensive antibody specific understanding and generation tasks. S$^2$ALM's ability to model comprehensive and generalized representations further positions its potential to advance real-world therapeutic antibody development, potentially addressing unmet academic, industrial, and clinical needs.
Multi-Modal CLIP-Informed Protein Editing
Jul 27, 2024Proteins govern most biological functions essential for life, but achieving controllable protein discovery and optimization remains challenging. Recently, machine learning-assisted protein editing (MLPE) has shown promise in accelerating optimization cycles and reducing experimental workloads. However, current methods struggle with the vast combinatorial space of potential protein edits and cannot explicitly conduct protein editing using biotext instructions, limiting their interactivity with human feedback. To fill these gaps, we propose a novel method called ProtET for efficient CLIP-informed protein editing through multi-modality learning. Our approach comprises two stages: in the pretraining stage, contrastive learning aligns protein-biotext representations encoded by two large language models (LLMs), respectively. Subsequently, during the protein editing stage, the fused features from editing instruction texts and original protein sequences serve as the final editing condition for generating target protein sequences. Comprehensive experiments demonstrated the superiority of ProtET in editing proteins to enhance human-expected functionality across multiple attribute domains, including enzyme catalytic activity, protein stability and antibody specific binding ability. And ProtET improves the state-of-the-art results by a large margin, leading to significant stability improvements of 16.67% and 16.90%. This capability positions ProtET to advance real-world artificial protein editing, potentially addressing unmet academic, industrial, and clinical needs.
Token-Mol 1.0: Tokenized drug design with large language model
Jul 10, 2024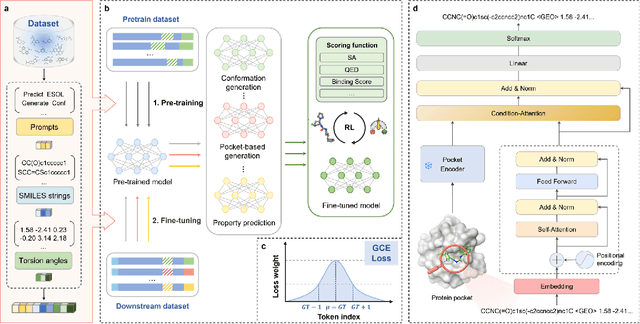

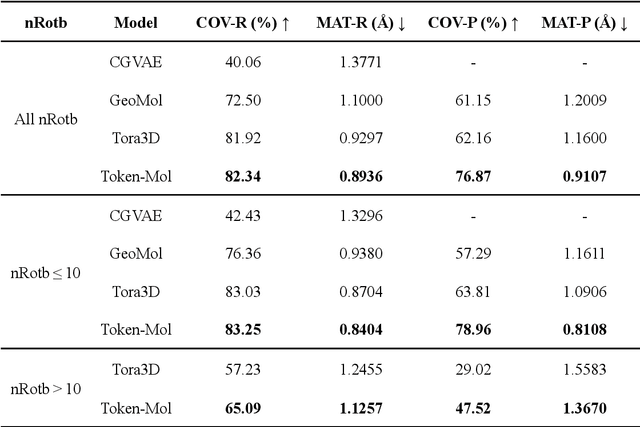
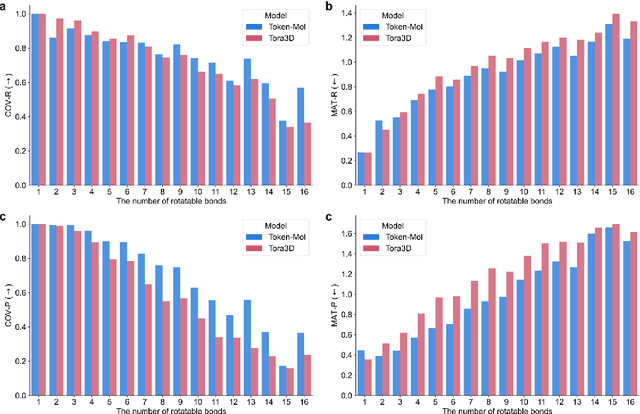
Significant interests have recently risen in leveraging sequence-based large language models (LLMs) for drug design. However, most current applications of LLMs in drug discovery lack the ability to comprehend three-dimensional (3D) structures, thereby limiting their effectiveness in tasks that explicitly involve molecular conformations. In this study, we introduced Token-Mol, a token-only 3D drug design model. This model encodes all molecular information, including 2D and 3D structures, as well as molecular property data, into tokens, which transforms classification and regression tasks in drug discovery into probabilistic prediction problems, thereby enabling learning through a unified paradigm. Token-Mol is built on the transformer decoder architecture and trained using random causal masking techniques. Additionally, we proposed the Gaussian cross-entropy (GCE) loss function to overcome the challenges in regression tasks, significantly enhancing the capacity of LLMs to learn continuous numerical values. Through a combination of fine-tuning and reinforcement learning (RL), Token-Mol achieves performance comparable to or surpassing existing task-specific methods across various downstream tasks, including pocket-based molecular generation, conformation generation, and molecular property prediction. Compared to existing molecular pre-trained models, Token-Mol exhibits superior proficiency in handling a wider range of downstream tasks essential for drug design. Notably, our approach improves regression task accuracy by approximately 30% compared to similar token-only methods. Token-Mol overcomes the precision limitations of token-only models and has the potential to integrate seamlessly with general models such as ChatGPT, paving the way for the development of a universal artificial intelligence drug design model that facilitates rapid and high-quality drug design by experts.
Deep Lead Optimization: Leveraging Generative AI for Structural Modification
Apr 30, 2024The idea of using deep-learning-based molecular generation to accelerate discovery of drug candidates has attracted extraordinary attention, and many deep generative models have been developed for automated drug design, termed molecular generation. In general, molecular generation encompasses two main strategies: de novo design, which generates novel molecular structures from scratch, and lead optimization, which refines existing molecules into drug candidates. Among them, lead optimization plays an important role in real-world drug design. For example, it can enable the development of me-better drugs that are chemically distinct yet more effective than the original drugs. It can also facilitate fragment-based drug design, transforming virtual-screened small ligands with low affinity into first-in-class medicines. Despite its importance, automated lead optimization remains underexplored compared to the well-established de novo generative models, due to its reliance on complex biological and chemical knowledge. To bridge this gap, we conduct a systematic review of traditional computational methods for lead optimization, organizing these strategies into four principal sub-tasks with defined inputs and outputs. This review delves into the basic concepts, goals, conventional CADD techniques, and recent advancements in AIDD. Additionally, we introduce a unified perspective based on constrained subgraph generation to harmonize the methodologies of de novo design and lead optimization. Through this lens, de novo design can incorporate strategies from lead optimization to address the challenge of generating hard-to-synthesize molecules; inversely, lead optimization can benefit from the innovations in de novo design by approaching it as a task of generating molecules conditioned on certain substructures.
AAVDiff: Experimental Validation of Enhanced Viability and Diversity in Recombinant Adeno-Associated Virus (AAV) Capsids through Diffusion Generation
Apr 17, 2024Recombinant adeno-associated virus (rAAV) vectors have revolutionized gene therapy, but their broad tropism and suboptimal transduction efficiency limit their clinical applications. To overcome these limitations, researchers have focused on designing and screening capsid libraries to identify improved vectors. However, the large sequence space and limited resources present challenges in identifying viable capsid variants. In this study, we propose an end-to-end diffusion model to generate capsid sequences with enhanced viability. Using publicly available AAV2 data, we generated 38,000 diverse AAV2 viral protein (VP) sequences, and evaluated 8,000 for viral selection. The results attested the superiority of our model compared to traditional methods. Additionally, in the absence of AAV9 capsid data, apart from one wild-type sequence, we used the same model to directly generate a number of viable sequences with up to 9 mutations. we transferred the remaining 30,000 samples to the AAV9 domain. Furthermore, we conducted mutagenesis on AAV9 VP hypervariable regions VI and V, contributing to the continuous improvement of the AAV9 VP sequence. This research represents a significant advancement in the design and functional validation of rAAV vectors, offering innovative solutions to enhance specificity and transduction efficiency in gene therapy applications.
Generative AI for Controllable Protein Sequence Design: A Survey
Feb 16, 2024

The design of novel protein sequences with targeted functionalities underpins a central theme in protein engineering, impacting diverse fields such as drug discovery and enzymatic engineering. However, navigating this vast combinatorial search space remains a severe challenge due to time and financial constraints. This scenario is rapidly evolving as the transformative advancements in AI, particularly in the realm of generative models and optimization algorithms, have been propelling the protein design field towards an unprecedented revolution. In this survey, we systematically review recent advances in generative AI for controllable protein sequence design. To set the stage, we first outline the foundational tasks in protein sequence design in terms of the constraints involved and present key generative models and optimization algorithms. We then offer in-depth reviews of each design task and discuss the pertinent applications. Finally, we identify the unresolved challenges and highlight research opportunities that merit deeper exploration.
Multiscale Topology in Interactomic Network: From Transcriptome to Antiaddiction Drug Repurposing
Dec 03, 2023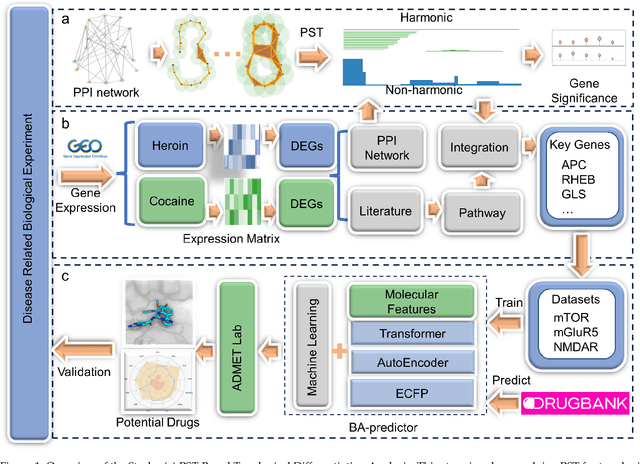
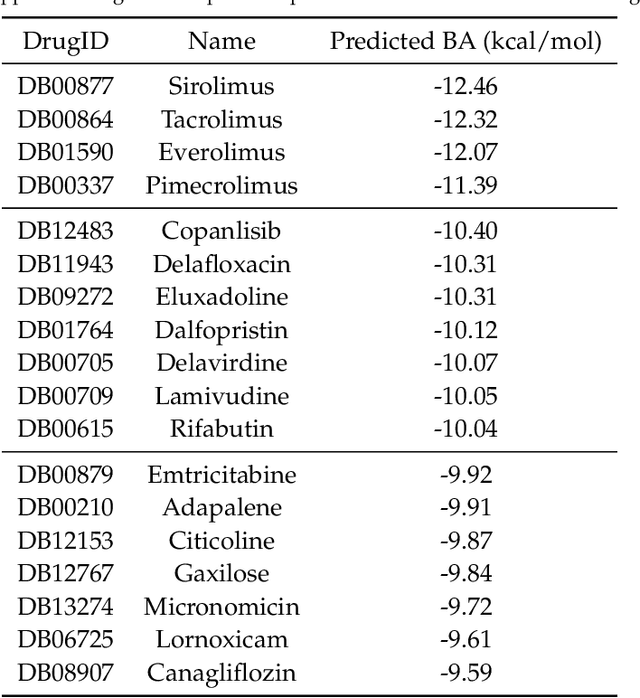
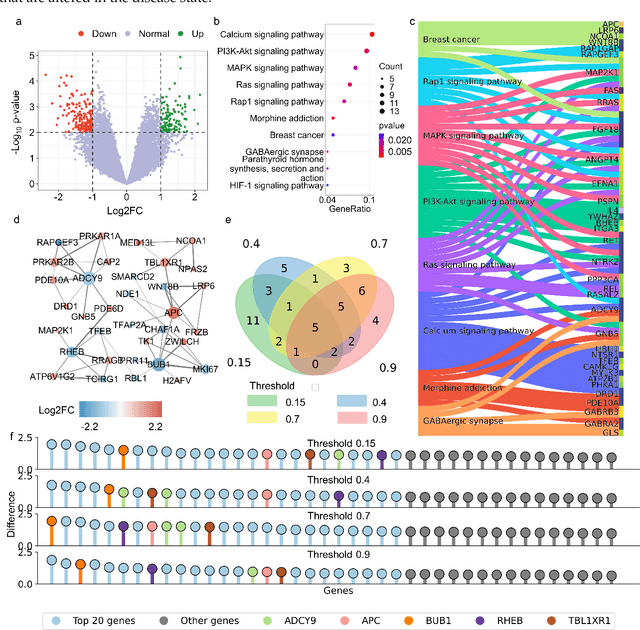
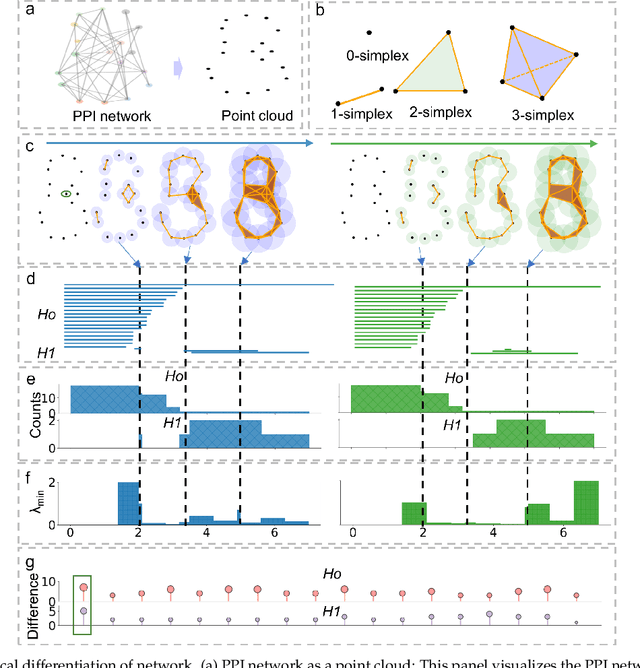
The escalating drug addiction crisis in the United States underscores the urgent need for innovative therapeutic strategies. This study embarked on an innovative and rigorous strategy to unearth potential drug repurposing candidates for opioid and cocaine addiction treatment, bridging the gap between transcriptomic data analysis and drug discovery. We initiated our approach by conducting differential gene expression analysis on addiction-related transcriptomic data to identify key genes. We propose a novel topological differentiation to identify key genes from a protein-protein interaction (PPI) network derived from DEGs. This method utilizes persistent Laplacians to accurately single out pivotal nodes within the network, conducting this analysis in a multiscale manner to ensure high reliability. Through rigorous literature validation, pathway analysis, and data-availability scrutiny, we identified three pivotal molecular targets, mTOR, mGluR5, and NMDAR, for drug repurposing from DrugBank. We crafted machine learning models employing two natural language processing (NLP)-based embeddings and a traditional 2D fingerprint, which demonstrated robust predictive ability in gauging binding affinities of DrugBank compounds to selected targets. Furthermore, we elucidated the interactions of promising drugs with the targets and evaluated their drug-likeness. This study delineates a multi-faceted and comprehensive analytical framework, amalgamating bioinformatics, topological data analysis and machine learning, for drug repurposing in addiction treatment, setting the stage for subsequent experimental validation. The versatility of the methods we developed allows for applications across a range of diseases and transcriptomic datasets.