 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeTesting Storage-System Correctness: Challenges, Fuzzing Limitations, and AI-Augmented Opportunities
Feb 02, 2026Storage systems are fundamental to modern computing infrastructures, yet ensuring their correctness remains challenging in practice. Despite decades of research on system testing, many storage-system failures (including durability, ordering, recovery, and consistency violations) remain difficult to expose systematically. This difficulty stems not primarily from insufficient testing tooling, but from intrinsic properties of storage-system execution, including nondeterministic interleavings, long-horizon state evolution, and correctness semantics that span multiple layers and execution phases. This survey adopts a storage-centric view of system testing and organizes existing techniques according to the execution properties and failure mechanisms they target. We review a broad spectrum of approaches, ranging from concurrency testing and long-running workloads to crash-consistency analysis, hardware-level semantic validation, and distributed fault injection, and analyze their fundamental strengths and limitations. Within this framework, we examine fuzzing as an automated testing paradigm, highlighting systematic mismatches between conventional fuzzing assumptions and storage-system semantics, and discuss how recent artificial intelligence advances may complement fuzzing through state-aware and semantic guidance. Overall, this survey provides a unified perspective on storage-system correctness testing and outlines key challenges
Token-Mol 1.0: Tokenized drug design with large language model
Jul 10, 2024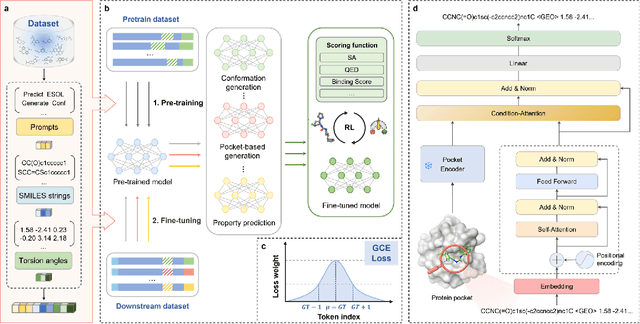

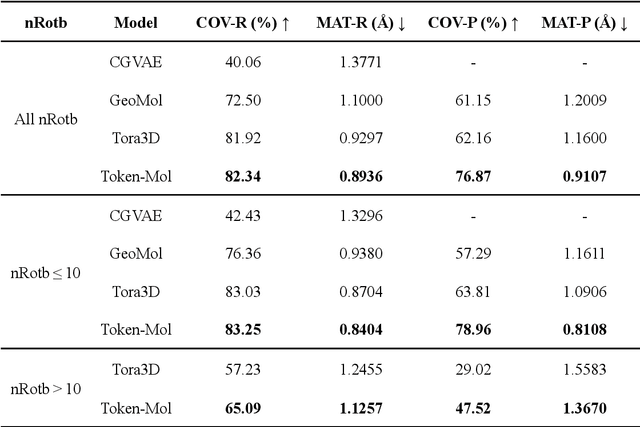
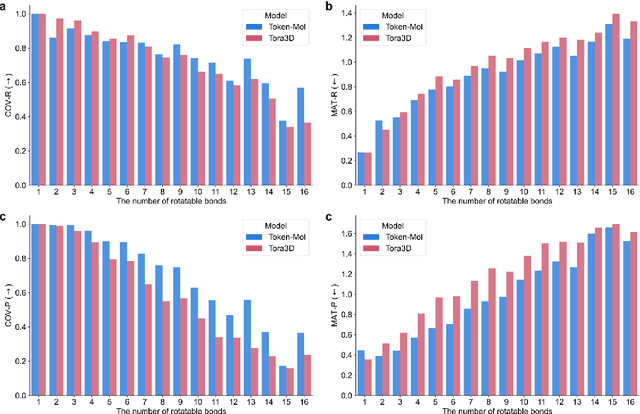
Significant interests have recently risen in leveraging sequence-based large language models (LLMs) for drug design. However, most current applications of LLMs in drug discovery lack the ability to comprehend three-dimensional (3D) structures, thereby limiting their effectiveness in tasks that explicitly involve molecular conformations. In this study, we introduced Token-Mol, a token-only 3D drug design model. This model encodes all molecular information, including 2D and 3D structures, as well as molecular property data, into tokens, which transforms classification and regression tasks in drug discovery into probabilistic prediction problems, thereby enabling learning through a unified paradigm. Token-Mol is built on the transformer decoder architecture and trained using random causal masking techniques. Additionally, we proposed the Gaussian cross-entropy (GCE) loss function to overcome the challenges in regression tasks, significantly enhancing the capacity of LLMs to learn continuous numerical values. Through a combination of fine-tuning and reinforcement learning (RL), Token-Mol achieves performance comparable to or surpassing existing task-specific methods across various downstream tasks, including pocket-based molecular generation, conformation generation, and molecular property prediction. Compared to existing molecular pre-trained models, Token-Mol exhibits superior proficiency in handling a wider range of downstream tasks essential for drug design. Notably, our approach improves regression task accuracy by approximately 30% compared to similar token-only methods. Token-Mol overcomes the precision limitations of token-only models and has the potential to integrate seamlessly with general models such as ChatGPT, paving the way for the development of a universal artificial intelligence drug design model that facilitates rapid and high-quality drug design by experts.
Deep Lead Optimization: Leveraging Generative AI for Structural Modification
Apr 30, 2024The idea of using deep-learning-based molecular generation to accelerate discovery of drug candidates has attracted extraordinary attention, and many deep generative models have been developed for automated drug design, termed molecular generation. In general, molecular generation encompasses two main strategies: de novo design, which generates novel molecular structures from scratch, and lead optimization, which refines existing molecules into drug candidates. Among them, lead optimization plays an important role in real-world drug design. For example, it can enable the development of me-better drugs that are chemically distinct yet more effective than the original drugs. It can also facilitate fragment-based drug design, transforming virtual-screened small ligands with low affinity into first-in-class medicines. Despite its importance, automated lead optimization remains underexplored compared to the well-established de novo generative models, due to its reliance on complex biological and chemical knowledge. To bridge this gap, we conduct a systematic review of traditional computational methods for lead optimization, organizing these strategies into four principal sub-tasks with defined inputs and outputs. This review delves into the basic concepts, goals, conventional CADD techniques, and recent advancements in AIDD. Additionally, we introduce a unified perspective based on constrained subgraph generation to harmonize the methodologies of de novo design and lead optimization. Through this lens, de novo design can incorporate strategies from lead optimization to address the challenge of generating hard-to-synthesize molecules; inversely, lead optimization can benefit from the innovations in de novo design by approaching it as a task of generating molecules conditioned on certain substructures.