 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGraph Neural Networks in Modern AI-aided Drug Discovery
Jun 07, 2025Graph neural networks (GNNs), as topology/structure-aware models within deep learning, have emerged as powerful tools for AI-aided drug discovery (AIDD). By directly operating on molecular graphs, GNNs offer an intuitive and expressive framework for learning the complex topological and geometric features of drug-like molecules, cementing their role in modern molecular modeling. This review provides a comprehensive overview of the methodological foundations and representative applications of GNNs in drug discovery, spanning tasks such as molecular property prediction, virtual screening, molecular generation, biomedical knowledge graph construction, and synthesis planning. Particular attention is given to recent methodological advances, including geometric GNNs, interpretable models, uncertainty quantification, scalable graph architectures, and graph generative frameworks. We also discuss how these models integrate with modern deep learning approaches, such as self-supervised learning, multi-task learning, meta-learning and pre-training. Throughout this review, we highlight the practical challenges and methodological bottlenecks encountered when applying GNNs to real-world drug discovery pipelines, and conclude with a discussion on future directions.
InstructBioMol: Advancing Biomolecule Understanding and Design Following Human Instructions
Oct 10, 2024Understanding and designing biomolecules, such as proteins and small molecules, is central to advancing drug discovery, synthetic biology, and enzyme engineering. Recent breakthroughs in Artificial Intelligence (AI) have revolutionized biomolecular research, achieving remarkable accuracy in biomolecular prediction and design. However, a critical gap remains between AI's computational power and researchers' intuition, using natural language to align molecular complexity with human intentions. Large Language Models (LLMs) have shown potential to interpret human intentions, yet their application to biomolecular research remains nascent due to challenges including specialized knowledge requirements, multimodal data integration, and semantic alignment between natural language and biomolecules. To address these limitations, we present InstructBioMol, a novel LLM designed to bridge natural language and biomolecules through a comprehensive any-to-any alignment of natural language, molecules, and proteins. This model can integrate multimodal biomolecules as input, and enable researchers to articulate design goals in natural language, providing biomolecular outputs that meet precise biological needs. Experimental results demonstrate InstructBioMol can understand and design biomolecules following human instructions. Notably, it can generate drug molecules with a 10% improvement in binding affinity and design enzymes that achieve an ESP Score of 70.4, making it the only method to surpass the enzyme-substrate interaction threshold of 60.0 recommended by the ESP developer. This highlights its potential to transform real-world biomolecular research.
Token-Mol 1.0: Tokenized drug design with large language model
Jul 10, 2024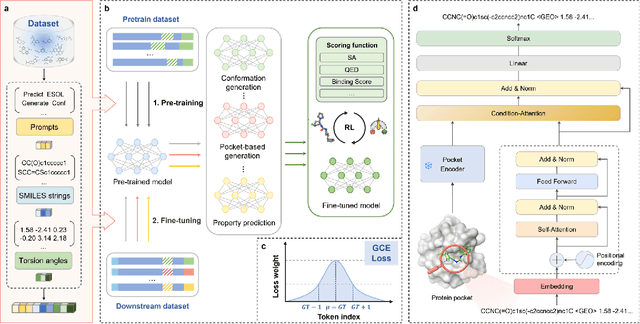

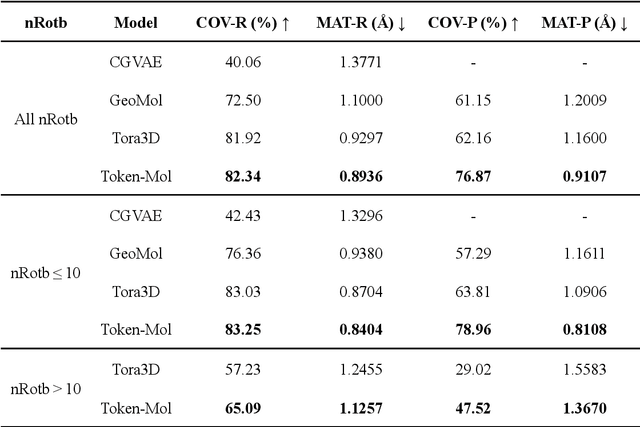
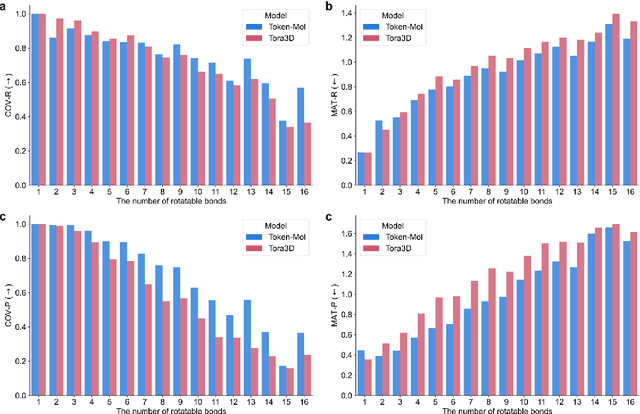
Significant interests have recently risen in leveraging sequence-based large language models (LLMs) for drug design. However, most current applications of LLMs in drug discovery lack the ability to comprehend three-dimensional (3D) structures, thereby limiting their effectiveness in tasks that explicitly involve molecular conformations. In this study, we introduced Token-Mol, a token-only 3D drug design model. This model encodes all molecular information, including 2D and 3D structures, as well as molecular property data, into tokens, which transforms classification and regression tasks in drug discovery into probabilistic prediction problems, thereby enabling learning through a unified paradigm. Token-Mol is built on the transformer decoder architecture and trained using random causal masking techniques. Additionally, we proposed the Gaussian cross-entropy (GCE) loss function to overcome the challenges in regression tasks, significantly enhancing the capacity of LLMs to learn continuous numerical values. Through a combination of fine-tuning and reinforcement learning (RL), Token-Mol achieves performance comparable to or surpassing existing task-specific methods across various downstream tasks, including pocket-based molecular generation, conformation generation, and molecular property prediction. Compared to existing molecular pre-trained models, Token-Mol exhibits superior proficiency in handling a wider range of downstream tasks essential for drug design. Notably, our approach improves regression task accuracy by approximately 30% compared to similar token-only methods. Token-Mol overcomes the precision limitations of token-only models and has the potential to integrate seamlessly with general models such as ChatGPT, paving the way for the development of a universal artificial intelligence drug design model that facilitates rapid and high-quality drug design by experts.