 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeOmni-Weather: Unified Multimodal Foundation Model for Weather Generation and Understanding
Dec 25, 2025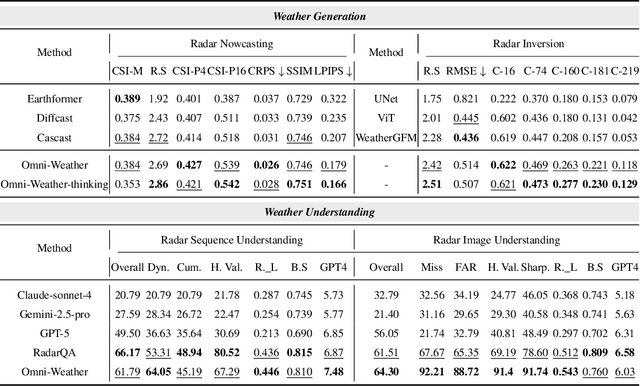
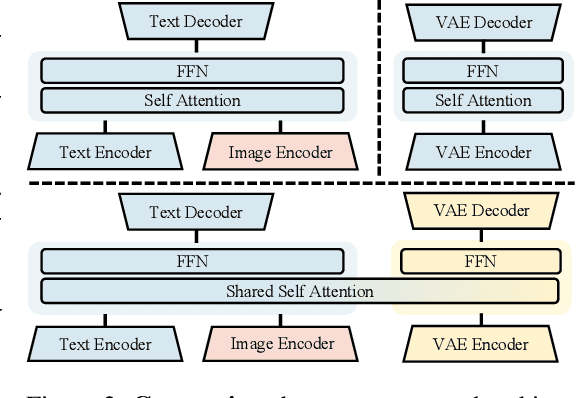
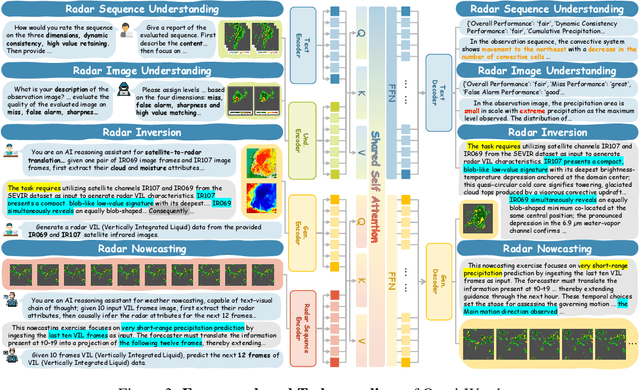

Weather modeling requires both accurate prediction and mechanistic interpretation, yet existing methods treat these goals in isolation, separating generation from understanding. To address this gap, we present Omni-Weather, the first multimodal foundation model that unifies weather generation and understanding within a single architecture. Omni-Weather integrates a radar encoder for weather generation tasks, followed by unified processing using a shared self-attention mechanism. Moreover, we construct a Chain-of-Thought dataset for causal reasoning in weather generation, enabling interpretable outputs and improved perceptual quality. Extensive experiments show Omni-Weather achieves state-of-the-art performance in both weather generation and understanding. Our findings further indicate that generative and understanding tasks in the weather domain can mutually enhance each other. Omni-Weather also demonstrates the feasibility and value of unifying weather generation and understanding.
Probing Scientific General Intelligence of LLMs with Scientist-Aligned Workflows
Dec 18, 2025Despite advances in scientific AI, a coherent framework for Scientific General Intelligence (SGI)-the ability to autonomously conceive, investigate, and reason across scientific domains-remains lacking. We present an operational SGI definition grounded in the Practical Inquiry Model (PIM: Deliberation, Conception, Action, Perception) and operationalize it via four scientist-aligned tasks: deep research, idea generation, dry/wet experiments, and experimental reasoning. SGI-Bench comprises over 1,000 expert-curated, cross-disciplinary samples inspired by Science's 125 Big Questions, enabling systematic evaluation of state-of-the-art LLMs. Results reveal gaps: low exact match (10--20%) in deep research despite step-level alignment; ideas lacking feasibility and detail; high code executability but low execution result accuracy in dry experiments; low sequence fidelity in wet protocols; and persistent multimodal comparative-reasoning challenges. We further introduce Test-Time Reinforcement Learning (TTRL), which optimizes retrieval-augmented novelty rewards at inference, enhancing hypothesis novelty without reference answer. Together, our PIM-grounded definition, workflow-centric benchmark, and empirical insights establish a foundation for AI systems that genuinely participate in scientific discovery.
Knowledge-Augmented Long-CoT Generation for Complex Biomolecular Reasoning
Nov 11, 2025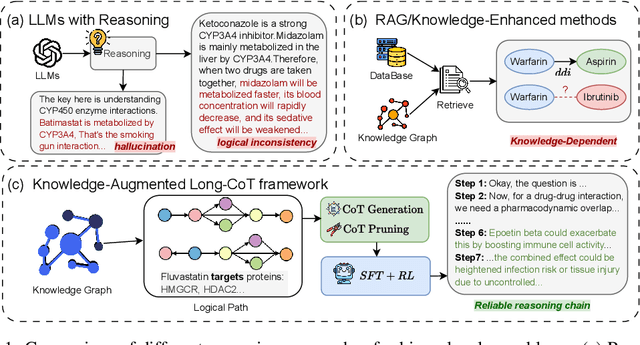
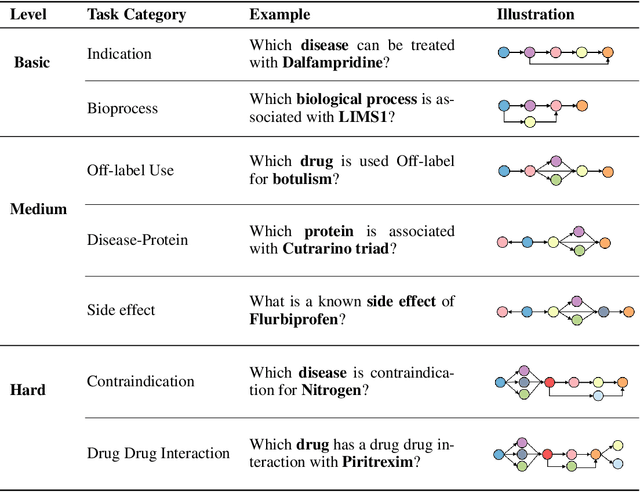
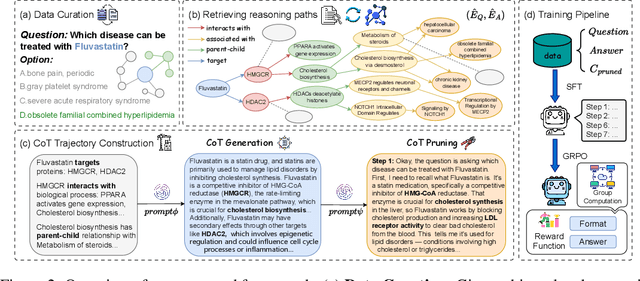
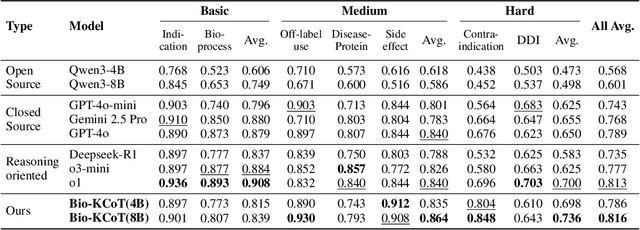
Understanding complex biomolecular mechanisms requires multi-step reasoning across molecular interactions, signaling cascades, and metabolic pathways. While large language models(LLMs) show promise in such tasks, their application to biomolecular problems is hindered by logical inconsistencies and the lack of grounding in domain knowledge. Existing approaches often exacerbate these issues: reasoning steps may deviate from biological facts or fail to capture long mechanistic dependencies. To address these challenges, we propose a Knowledge-Augmented Long-CoT Reasoning framework that integrates LLMs with knowledge graph-based multi-hop reasoning chains. The framework constructs mechanistic chains via guided multi-hop traversal and pruning on the knowledge graph; these chains are then incorporated into supervised fine-tuning to improve factual grounding and further refined with reinforcement learning to enhance reasoning reliability and consistency. Furthermore, to overcome the shortcomings of existing benchmarks, which are often restricted in scale and scope and lack annotations for deep reasoning chains, we introduce PrimeKGQA, a comprehensive benchmark for biomolecular question answering. Experimental results on both PrimeKGQA and existing datasets demonstrate that although larger closed-source models still perform well on relatively simple tasks, our method demonstrates clear advantages as reasoning depth increases, achieving state-of-the-art performance on multi-hop tasks that demand traversal of structured biological knowledge. These findings highlight the effectiveness of combining structured knowledge with advanced reasoning strategies for reliable and interpretable biomolecular reasoning.
Breaking the Modality Barrier: Generative Modeling for Accurate Molecule Retrieval from Mass Spectra
Nov 09, 2025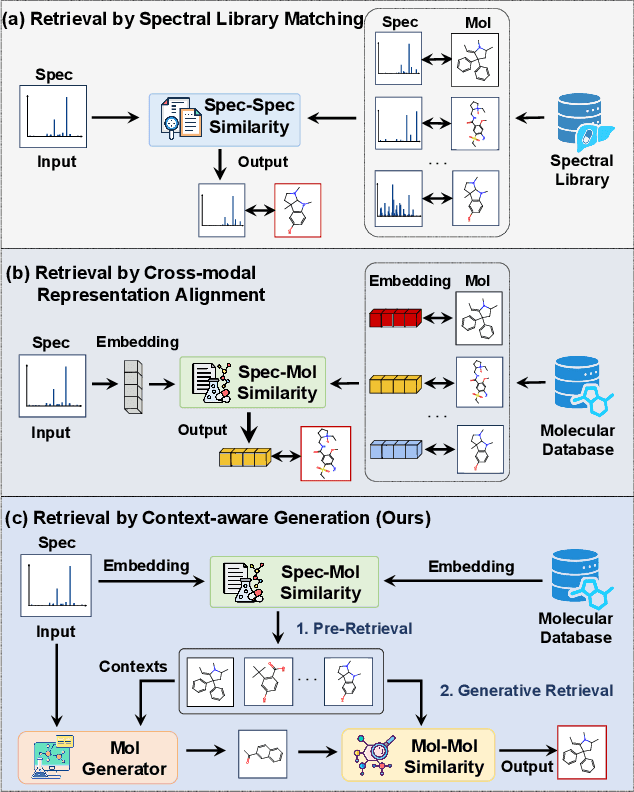
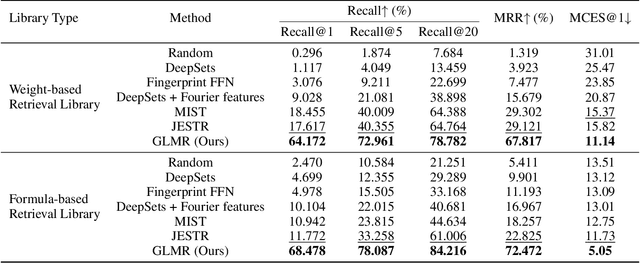
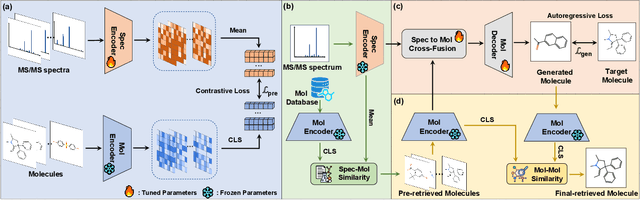
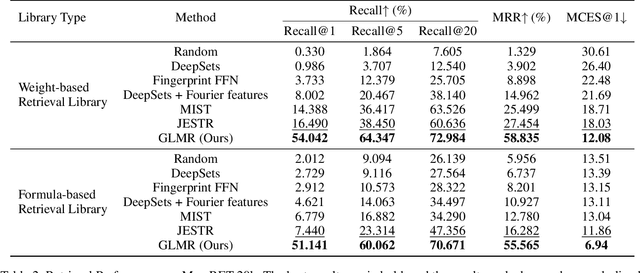
Retrieving molecular structures from tandem mass spectra is a crucial step in rapid compound identification. Existing retrieval methods, such as traditional mass spectral library matching, suffer from limited spectral library coverage, while recent cross-modal representation learning frameworks often encounter modality misalignment, resulting in suboptimal retrieval accuracy and generalization. To address these limitations, we propose GLMR, a Generative Language Model-based Retrieval framework that mitigates the cross-modal misalignment through a two-stage process. In the pre-retrieval stage, a contrastive learning-based model identifies top candidate molecules as contextual priors for the input mass spectrum. In the generative retrieval stage, these candidate molecules are integrated with the input mass spectrum to guide a generative model in producing refined molecular structures, which are then used to re-rank the candidates based on molecular similarity. Experiments on both MassSpecGym and the proposed MassRET-20k dataset demonstrate that GLMR significantly outperforms existing methods, achieving over 40% improvement in top-1 accuracy and exhibiting strong generalizability.
InstructBioMol: Advancing Biomolecule Understanding and Design Following Human Instructions
Oct 10, 2024Understanding and designing biomolecules, such as proteins and small molecules, is central to advancing drug discovery, synthetic biology, and enzyme engineering. Recent breakthroughs in Artificial Intelligence (AI) have revolutionized biomolecular research, achieving remarkable accuracy in biomolecular prediction and design. However, a critical gap remains between AI's computational power and researchers' intuition, using natural language to align molecular complexity with human intentions. Large Language Models (LLMs) have shown potential to interpret human intentions, yet their application to biomolecular research remains nascent due to challenges including specialized knowledge requirements, multimodal data integration, and semantic alignment between natural language and biomolecules. To address these limitations, we present InstructBioMol, a novel LLM designed to bridge natural language and biomolecules through a comprehensive any-to-any alignment of natural language, molecules, and proteins. This model can integrate multimodal biomolecules as input, and enable researchers to articulate design goals in natural language, providing biomolecular outputs that meet precise biological needs. Experimental results demonstrate InstructBioMol can understand and design biomolecules following human instructions. Notably, it can generate drug molecules with a 10% improvement in binding affinity and design enzymes that achieve an ESP Score of 70.4, making it the only method to surpass the enzyme-substrate interaction threshold of 60.0 recommended by the ESP developer. This highlights its potential to transform real-world biomolecular research.
SciKnowEval: Evaluating Multi-level Scientific Knowledge of Large Language Models
Jun 13, 2024The burgeoning utilization of Large Language Models (LLMs) in scientific research necessitates advanced benchmarks capable of evaluating their understanding and application of scientific knowledge comprehensively. To address this need, we introduce the SciKnowEval benchmark, a novel framework that systematically evaluates LLMs across five progressive levels of scientific knowledge: studying extensively, inquiring earnestly, thinking profoundly, discerning clearly, and practicing assiduously. These levels aim to assess the breadth and depth of scientific knowledge in LLMs, including knowledge coverage, inquiry and exploration capabilities, reflection and reasoning abilities, ethic and safety considerations, as well as practice proficiency. Specifically, we take biology and chemistry as the two instances of SciKnowEval and construct a dataset encompassing 50K multi-level scientific problems and solutions. By leveraging this dataset, we benchmark 20 leading open-source and proprietary LLMs using zero-shot and few-shot prompting strategies. The results reveal that despite achieving state-of-the-art performance, the proprietary LLMs still have considerable room for improvement, particularly in addressing scientific computations and applications. We anticipate that SciKnowEval will establish a comprehensive standard for benchmarking LLMs in science research and discovery, and promote the development of LLMs that integrate scientific knowledge with strong safety awareness. The dataset and code are publicly available at https://github.com/hicai-zju/sciknoweval .
StableMask: Refining Causal Masking in Decoder-only Transformer
Feb 07, 2024



The decoder-only Transformer architecture with causal masking and relative position encoding (RPE) has become the de facto choice in language modeling. Despite its exceptional performance across various tasks, we have identified two limitations: First, it requires all attention scores to be non-zero and sum up to 1, even if the current embedding has sufficient self-contained information. This compels the model to assign disproportional excessive attention to specific tokens. Second, RPE-based Transformers are not universal approximators due to their limited capacity at encoding absolute positional information, which limits their application in position-critical tasks. In this work, we propose StableMask: a parameter-free method to address both limitations by refining the causal mask. It introduces pseudo-attention values to balance attention distributions and encodes absolute positional information via a progressively decreasing mask ratio. StableMask's effectiveness is validated both theoretically and empirically, showing significant enhancements in language models with parameter sizes ranging from 71M to 1.4B across diverse datasets and encoding methods. We further show that it naturally supports (1) efficient extrapolation without special tricks such as StreamingLLM and (2) easy integration with existing attention optimization techniques.
Scientific Large Language Models: A Survey on Biological & Chemical Domains
Jan 26, 2024
Large Language Models (LLMs) have emerged as a transformative power in enhancing natural language comprehension, representing a significant stride toward artificial general intelligence. The application of LLMs extends beyond conventional linguistic boundaries, encompassing specialized linguistic systems developed within various scientific disciplines. This growing interest has led to the advent of scientific LLMs, a novel subclass specifically engineered for facilitating scientific discovery. As a burgeoning area in the community of AI for Science, scientific LLMs warrant comprehensive exploration. However, a systematic and up-to-date survey introducing them is currently lacking. In this paper, we endeavor to methodically delineate the concept of "scientific language", whilst providing a thorough review of the latest advancements in scientific LLMs. Given the expansive realm of scientific disciplines, our analysis adopts a focused lens, concentrating on the biological and chemical domains. This includes an in-depth examination of LLMs for textual knowledge, small molecules, macromolecular proteins, genomic sequences, and their combinations, analyzing them in terms of model architectures, capabilities, datasets, and evaluation. Finally, we critically examine the prevailing challenges and point out promising research directions along with the advances of LLMs. By offering a comprehensive overview of technical developments in this field, this survey aspires to be an invaluable resource for researchers navigating the intricate landscape of scientific LLMs.
Learning Invariant Molecular Representation in Latent Discrete Space
Oct 22, 2023Molecular representation learning lays the foundation for drug discovery. However, existing methods suffer from poor out-of-distribution (OOD) generalization, particularly when data for training and testing originate from different environments. To address this issue, we propose a new framework for learning molecular representations that exhibit invariance and robustness against distribution shifts. Specifically, we propose a strategy called ``first-encoding-then-separation'' to identify invariant molecule features in the latent space, which deviates from conventional practices. Prior to the separation step, we introduce a residual vector quantization module that mitigates the over-fitting to training data distributions while preserving the expressivity of encoders. Furthermore, we design a task-agnostic self-supervised learning objective to encourage precise invariance identification, which enables our method widely applicable to a variety of tasks, such as regression and multi-label classification. Extensive experiments on 18 real-world molecular datasets demonstrate that our model achieves stronger generalization against state-of-the-art baselines in the presence of various distribution shifts. Our code is available at https://github.com/HICAI-ZJU/iMoLD.
InstructProtein: Aligning Human and Protein Language via Knowledge Instruction
Oct 05, 2023Large Language Models (LLMs) have revolutionized the field of natural language processing, but they fall short in comprehending biological sequences such as proteins. To address this challenge, we propose InstructProtein, an innovative LLM that possesses bidirectional generation capabilities in both human and protein languages: (i) taking a protein sequence as input to predict its textual function description and (ii) using natural language to prompt protein sequence generation. To achieve this, we first pre-train an LLM on both protein and natural language corpora, enabling it to comprehend individual languages. Then supervised instruction tuning is employed to facilitate the alignment of these two distinct languages. Herein, we introduce a knowledge graph-based instruction generation framework to construct a high-quality instruction dataset, addressing annotation imbalance and instruction deficits in existing protein-text corpus. In particular, the instructions inherit the structural relations between proteins and function annotations in knowledge graphs, which empowers our model to engage in the causal modeling of protein functions, akin to the chain-of-thought processes in natural languages. Extensive experiments on bidirectional protein-text generation tasks show that InstructProtein outperforms state-of-the-art LLMs by large margins. Moreover, InstructProtein serves as a pioneering step towards text-based protein function prediction and sequence design, effectively bridging the gap between protein and human language understanding.