 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeGeneric Interpretation Approach for Transformer Models Incorporating Heterogenous Attention Structures
May 25, 2026Transformer has significantly propelled the development of artificial intelligence, and certainly the development of agents as well. We categorize attention structures of Transformer into two types based on the source of the input information: homogenous and heterogenous attention structures. Heterogenous attention structures, with co-attention as a typical example, process information from different sources. Heterogenous attention structure is the foundation for Transformer models to achieve more complex functions and integrate more modal information. Whether for research purposes or policy requirements, the interpretation of Transformer models with heterogenous attention structures is an important task. The fusion of information from different sources brings new challenges. Our work mainly includes two parts: method and experimentation. In terms of method, we propose an interpretation method for Transformer models with heterogenous attention structures. In terms of experimentation, based on our experimental analysis paradigm, we interpret the operating mechanisms of representative models, conduct semantic interpretation and logical interpretation.
The Neglected Baseline in Model Interpretation
May 21, 2026We observe that existing model interpretation methods generally ignore the baseline, and such neglect often results in imprecise or even incorrect interpretation. In this paper, we reformulate the task of model interpretation and the interpretation principles for model interpretation results to demonstrate the importance of the baseline. We further unify gradient-based methods, Integrated Gradients (IG) methods, and Taylor expansion, clarifying the connections among them and explicitly identifying the baseline for each method. On this basis, we analyze the flaws and errors in related model interpretation methods (IG, LayerCAM, ODAM, Difference Map). We advocate evaluating the quality of model interpretation results precisely through the attribution error between the attribution result and the attribution target, rather than adopting flawed evaluation methods, such as those based on marginal-effect or the assumption of perfect model performance. We revise IG and develope a model interpretation method with a clear and reasonable baseline, achieving better results. Our method supports model interpretation based on features from any layer. Interpretation based on features from different layers are all reasonable, and the differences among these results reflect varying degrees of feature extraction at different feature extraction stages.
Debunking Grad-ECLIP: A Comprehensive Study on Its Incorrectness and Fundamental Principles for Model Interpretation
May 13, 2026Grad-ECLIP is published at ICML 2024 and represents a new Transformer interpretation technical route (intermediate features-based). First, this paper demonstrates that the intermediate features-based technical route is not a novel one. Based on the existing attention-based route, we have developed Attention-ECLIP, which is completely equivalent to Grad-ECLIP but with simpler computation. Both through formal derivation and experimental validation, we prove that the intermediate feature-based route represented by Grad-ECLIP is actually an equivalent variant of the attention-based route. Next, this paper demonstrates that the Grad-ECLIP method is flawed. The model interpretation results obtained by Grad-ECLIP are not those of the original model, and the interpretation results are misaligned with the model's performance. We analyze the causes of Grad-ECLIP's flaws and propose, or rather, explicitly emphasize two fundamental principles that model interpretation should adhere to in order to avoid similar errors.
PCIE_Pose Solution for EgoExo4D Pose and Proficiency Estimation Challenge
May 30, 2025This report introduces our team's (PCIE_EgoPose) solutions for the EgoExo4D Pose and Proficiency Estimation Challenges at CVPR2025. Focused on the intricate task of estimating 21 3D hand joints from RGB egocentric videos, which are complicated by subtle movements and frequent occlusions, we developed the Hand Pose Vision Transformer (HP-ViT+). This architecture synergizes a Vision Transformer and a CNN backbone, using weighted fusion to refine the hand pose predictions. For the EgoExo4D Body Pose Challenge, we adopted a multimodal spatio-temporal feature integration strategy to address the complexities of body pose estimation across dynamic contexts. Our methods achieved remarkable performance: 8.31 PA-MPJPE in the Hand Pose Challenge and 11.25 MPJPE in the Body Pose Challenge, securing championship titles in both competitions. We extended our pose estimation solutions to the Proficiency Estimation task, applying core technologies such as transformer-based architectures. This extension enabled us to achieve a top-1 accuracy of 0.53, a SOTA result, in the Demonstrator Proficiency Estimation competition.
Ordered Genetic Algorithm for Entrance Dependent Vehicle Routing Problem in Farms
Feb 26, 2025Vehicle Routing Problems (VRP) are widely studied issues that play important roles in many production scenarios. We have noticed that in some practical scenarios of VRP, the size of cities and their entrances can significantly influence the optimization process. To address this, we have constructed the Entrance Dependent VRP (EDVRP) to describe such problems. We provide a mathematical formulation for the EDVRP in farms and propose an Ordered Genetic Algorithm (OGA) to solve it. The effectiveness of OGA is demonstrated through our experiments, which involve a multitude of randomly generated cases. The results indicate that OGA offers certain advantages compared to a random strategy baseline and a genetic algorithm without ordering. Furthermore, the novel operators introduced in this paper have been validated through ablation experiments, proving their effectiveness in enhancing the performance of the algorithm.
A Multi-Modal AI Copilot for Single-Cell Analysis with Instruction Following
Jan 15, 2025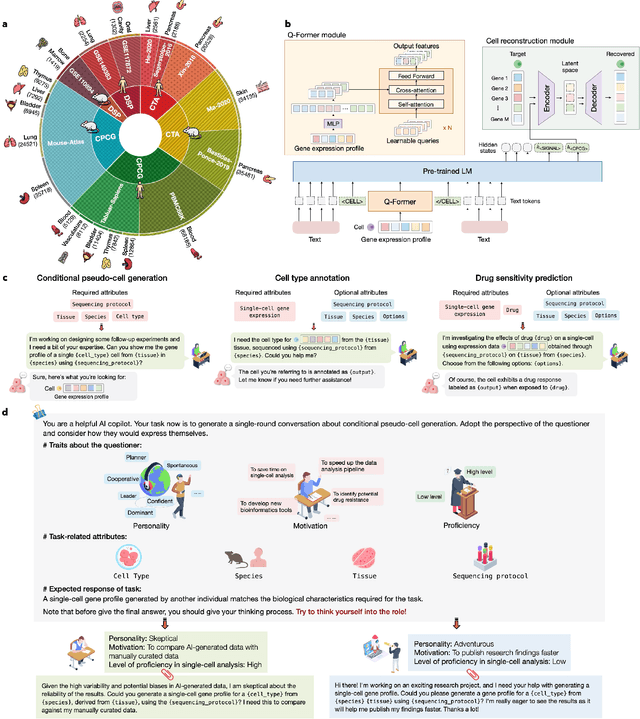
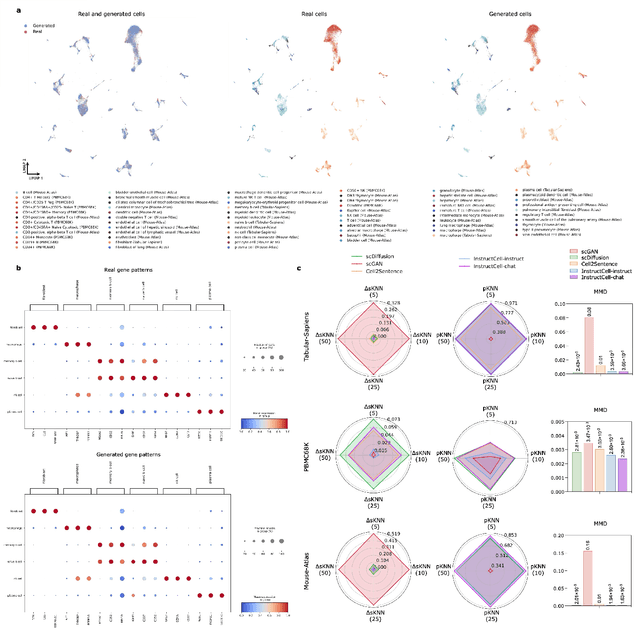
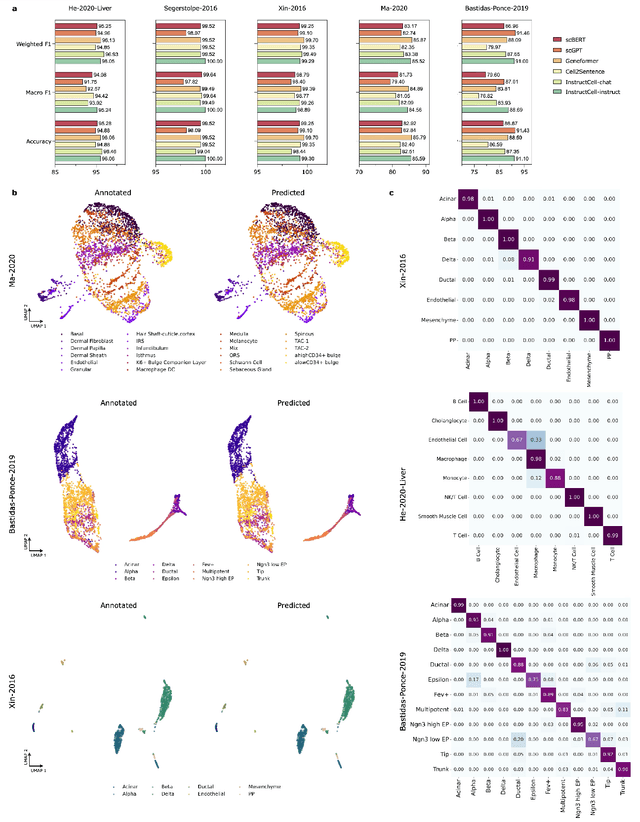
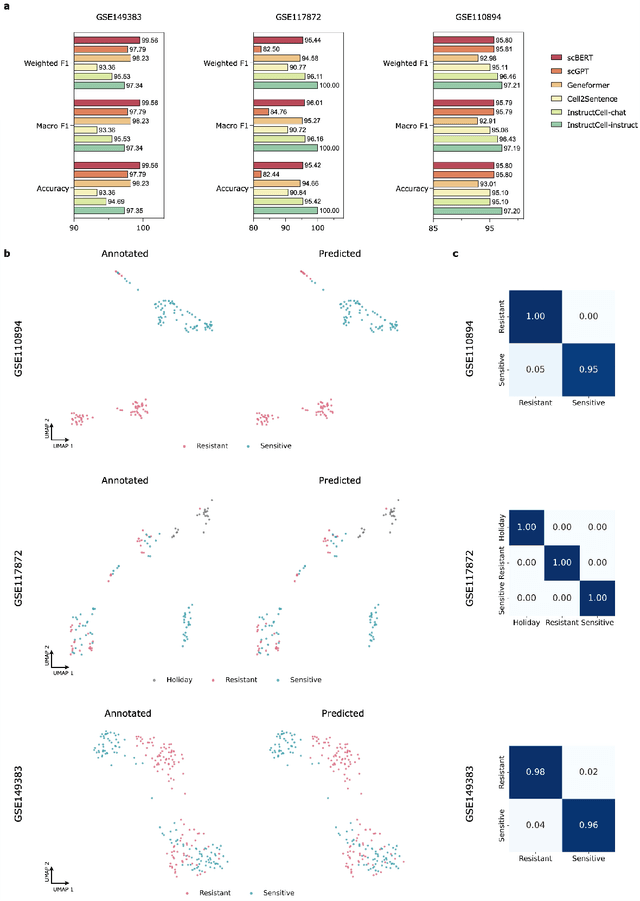
Large language models excel at interpreting complex natural language instructions, enabling them to perform a wide range of tasks. In the life sciences, single-cell RNA sequencing (scRNA-seq) data serves as the "language of cellular biology", capturing intricate gene expression patterns at the single-cell level. However, interacting with this "language" through conventional tools is often inefficient and unintuitive, posing challenges for researchers. To address these limitations, we present InstructCell, a multi-modal AI copilot that leverages natural language as a medium for more direct and flexible single-cell analysis. We construct a comprehensive multi-modal instruction dataset that pairs text-based instructions with scRNA-seq profiles from diverse tissues and species. Building on this, we develop a multi-modal cell language architecture capable of simultaneously interpreting and processing both modalities. InstructCell empowers researchers to accomplish critical tasks-such as cell type annotation, conditional pseudo-cell generation, and drug sensitivity prediction-using straightforward natural language commands. Extensive evaluations demonstrate that InstructCell consistently meets or exceeds the performance of existing single-cell foundation models, while adapting to diverse experimental conditions. More importantly, InstructCell provides an accessible and intuitive tool for exploring complex single-cell data, lowering technical barriers and enabling deeper biological insights.
ChatCell: Facilitating Single-Cell Analysis with Natural Language
Feb 20, 2024



As Large Language Models (LLMs) rapidly evolve, their influence in science is becoming increasingly prominent. The emerging capabilities of LLMs in task generalization and free-form dialogue can significantly advance fields like chemistry and biology. However, the field of single-cell biology, which forms the foundational building blocks of living organisms, still faces several challenges. High knowledge barriers and limited scalability in current methods restrict the full exploitation of LLMs in mastering single-cell data, impeding direct accessibility and rapid iteration. To this end, we introduce ChatCell, which signifies a paradigm shift by facilitating single-cell analysis with natural language. Leveraging vocabulary adaptation and unified sequence generation, ChatCell has acquired profound expertise in single-cell biology and the capability to accommodate a diverse range of analysis tasks. Extensive experiments further demonstrate ChatCell's robust performance and potential to deepen single-cell insights, paving the way for more accessible and intuitive exploration in this pivotal field. Our project homepage is available at https://zjunlp.github.io/project/ChatCell.
Scientific Large Language Models: A Survey on Biological & Chemical Domains
Jan 26, 2024
Large Language Models (LLMs) have emerged as a transformative power in enhancing natural language comprehension, representing a significant stride toward artificial general intelligence. The application of LLMs extends beyond conventional linguistic boundaries, encompassing specialized linguistic systems developed within various scientific disciplines. This growing interest has led to the advent of scientific LLMs, a novel subclass specifically engineered for facilitating scientific discovery. As a burgeoning area in the community of AI for Science, scientific LLMs warrant comprehensive exploration. However, a systematic and up-to-date survey introducing them is currently lacking. In this paper, we endeavor to methodically delineate the concept of "scientific language", whilst providing a thorough review of the latest advancements in scientific LLMs. Given the expansive realm of scientific disciplines, our analysis adopts a focused lens, concentrating on the biological and chemical domains. This includes an in-depth examination of LLMs for textual knowledge, small molecules, macromolecular proteins, genomic sequences, and their combinations, analyzing them in terms of model architectures, capabilities, datasets, and evaluation. Finally, we critically examine the prevailing challenges and point out promising research directions along with the advances of LLMs. By offering a comprehensive overview of technical developments in this field, this survey aspires to be an invaluable resource for researchers navigating the intricate landscape of scientific LLMs.
Mol-Instructions: A Large-Scale Biomolecular Instruction Dataset for Large Language Models
Jun 13, 2023


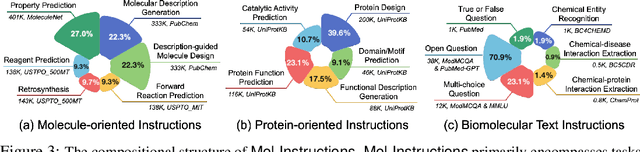
Large Language Models (LLMs), with their remarkable task-handling capabilities and innovative outputs, have catalyzed significant advancements across a spectrum of fields. However, their proficiency within specialized domains such as biomolecular studies remains limited. To address this challenge, we introduce Mol-Instructions, a meticulously curated, comprehensive instruction dataset expressly designed for the biomolecular realm. Mol-Instructions is composed of three pivotal components: molecule-oriented instructions, protein-oriented instructions, and biomolecular text instructions, each curated to enhance the understanding and prediction capabilities of LLMs concerning biomolecular features and behaviors. Through extensive instruction tuning experiments on the representative LLM, we underscore the potency of Mol-Instructions to enhance the adaptability and cognitive acuity of large models within the complex sphere of biomolecular studies, thereby promoting advancements in the biomolecular research community. Mol-Instructions is made publicly accessible for future research endeavors and will be subjected to continual updates for enhanced applicability.
Molecular Language Model as Multi-task Generator
Jan 29, 2023
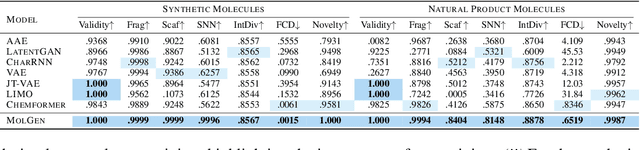
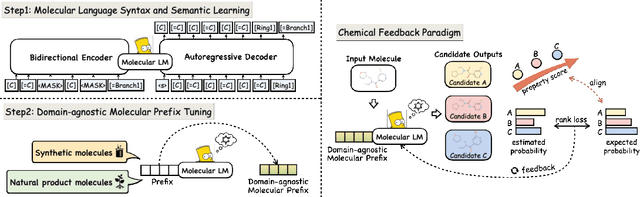
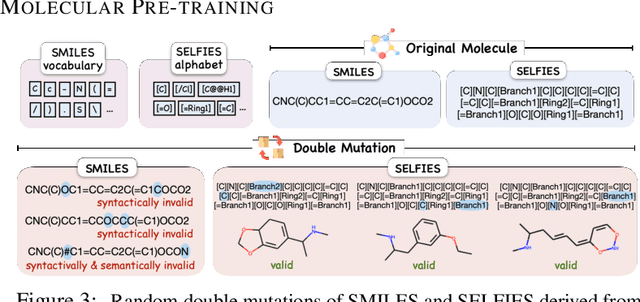
Molecule generation with desired properties has grown immensely in popularity by disruptively changing the way scientists design molecular structures and providing support for chemical and materials design. However, despite the promising outcome, previous machine learning-based deep generative models suffer from a reliance on complex, task-specific fine-tuning, limited dimensional latent spaces, or the quality of expert rules. In this work, we propose MolGen, a pre-trained molecular language model that effectively learns and shares knowledge across multiple generation tasks and domains. Specifically, we pre-train MolGen with the chemical language SELFIES on more than 100 million unlabelled molecules. We further propose multi-task molecular prefix tuning across several molecular generation tasks and different molecular domains (synthetic & natural products) with a self-feedback mechanism. Extensive experiments show that MolGen can obtain superior performances on well-known molecular generation benchmark datasets. The further analysis illustrates that MolGen can accurately capture the distribution of molecules, implicitly learn their structural characteristics, and efficiently explore the chemical space with the guidance of multi-task molecular prefix tuning. Codes, datasets, and the pre-trained model will be available in https://github.com/zjunlp/MolGen.