 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeApplication of Deep Generative Models for Anomaly Detection in Complex Financial Transactions
Apr 21, 2025This study proposes an algorithm for detecting suspicious behaviors in large payment flows based on deep generative models. By combining Generative Adversarial Networks (GAN) and Variational Autoencoders (VAE), the algorithm is designed to detect abnormal behaviors in financial transactions. First, the GAN is used to generate simulated data that approximates normal payment flows. The discriminator identifies anomalous patterns in transactions, enabling the detection of potential fraud and money laundering behaviors. Second, a VAE is introduced to model the latent distribution of payment flows, ensuring that the generated data more closely resembles real transaction features, thus improving the model's detection accuracy. The method optimizes the generative capabilities of both GAN and VAE, ensuring that the model can effectively capture suspicious behaviors even in sparse data conditions. Experimental results show that the proposed method significantly outperforms traditional machine learning algorithms and other deep learning models across various evaluation metrics, especially in detecting rare fraudulent behaviors. Furthermore, this study provides a detailed comparison of performance in recognizing different transaction patterns (such as normal, money laundering, and fraud) in large payment flows, validating the advantages of generative models in handling complex financial data.
Strategic priorities for transformative progress in advancing biology with proteomics and artificial intelligence
Feb 21, 2025

Artificial intelligence (AI) is transforming scientific research, including proteomics. Advances in mass spectrometry (MS)-based proteomics data quality, diversity, and scale, combined with groundbreaking AI techniques, are unlocking new challenges and opportunities in biological discovery. Here, we highlight key areas where AI is driving innovation, from data analysis to new biological insights. These include developing an AI-friendly ecosystem for proteomics data generation, sharing, and analysis; improving peptide and protein identification and quantification; characterizing protein-protein interactions and protein complexes; advancing spatial and perturbation proteomics; integrating multi-omics data; and ultimately enabling AI-empowered virtual cells.
Stock Type Prediction Model Based on Hierarchical Graph Neural Network
Dec 09, 2024

This paper introduces a novel approach to stock data analysis by employing a Hierarchical Graph Neural Network (HGNN) model that captures multi-level information and relational structures in the stock market. The HGNN model integrates stock relationship data and hierarchical attributes to predict stock types effectively. The paper discusses the construction of a stock industry relationship graph and the extraction of temporal information from historical price sequences. It also highlights the design of a graph convolution operation and a temporal attention aggregator to model the macro market state. The integration of these features results in a comprehensive stock prediction model that addresses the challenges of utilizing stock relationship data and modeling hierarchical attributes in the stock market.
Dynamic Fraud Detection: Integrating Reinforcement Learning into Graph Neural Networks
Sep 15, 2024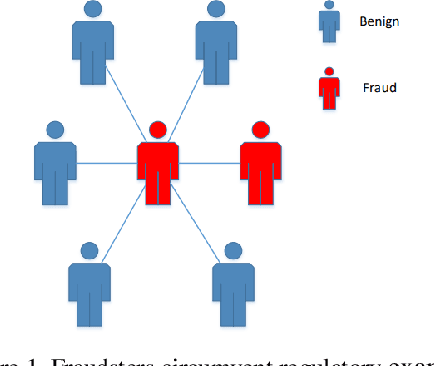
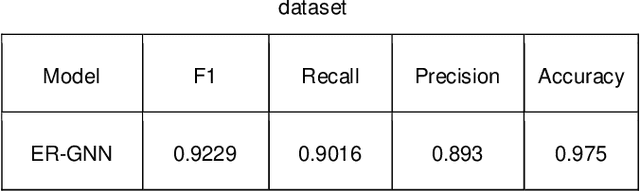
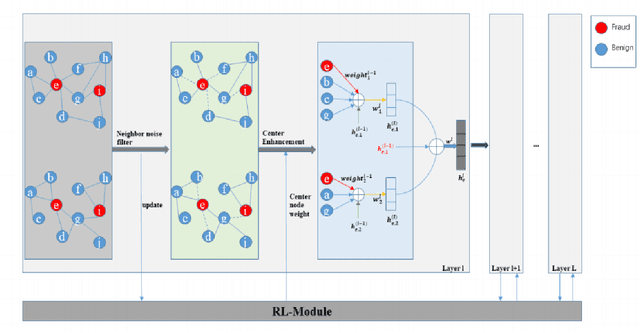
Financial fraud refers to the act of obtaining financial benefits through dishonest means. Such behavior not only disrupts the order of the financial market but also harms economic and social development and breeds other illegal and criminal activities. With the popularization of the internet and online payment methods, many fraudulent activities and money laundering behaviors in life have shifted from offline to online, posing a great challenge to regulatory authorities. How to efficiently detect these financial fraud activities has become an urgent issue that needs to be resolved. Graph neural networks are a type of deep learning model that can utilize the interactive relationships within graph structures, and they have been widely applied in the field of fraud detection. However, there are still some issues. First, fraudulent activities only account for a very small part of transaction transfers, leading to an inevitable problem of label imbalance in fraud detection. At the same time, fraudsters often disguise their behavior, which can have a negative impact on the final prediction results. In addition, existing research has overlooked the importance of balancing neighbor information and central node information. For example, when the central node has too many neighbors, the features of the central node itself are often neglected. Finally, fraud activities and patterns are constantly changing over time, so considering the dynamic evolution of graph edge relationships is also very important.
Meta-Learn Unimodal Signals with Weak Supervision for Multimodal Sentiment Analysis
Aug 28, 2024
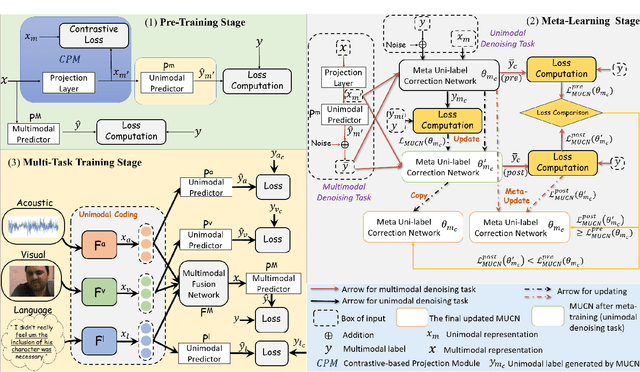
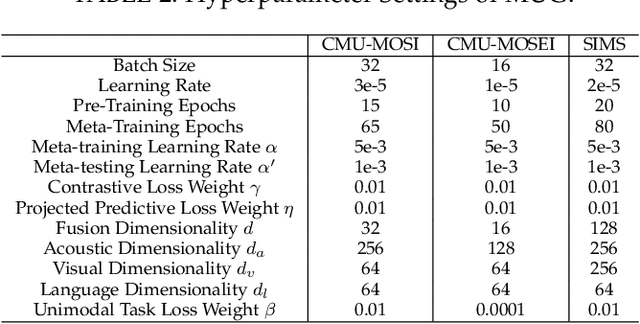
Multimodal sentiment analysis aims to effectively integrate information from various sources to infer sentiment, where in many cases there are no annotations for unimodal labels. Therefore, most works rely on multimodal labels for training. However, there exists the noisy label problem for the learning of unimodal signals as multimodal annotations are not always the ideal substitutes for the unimodal ones, failing to achieve finer optimization for individual modalities. In this paper, we explore the learning of unimodal labels under the weak supervision from the annotated multimodal labels. Specifically, we propose a novel meta uni-label generation (MUG) framework to address the above problem, which leverages the available multimodal labels to learn the corresponding unimodal labels by the meta uni-label correction network (MUCN). We first design a contrastive-based projection module to bridge the gap between unimodal and multimodal representations, so as to use multimodal annotations to guide the learning of MUCN. Afterwards, we propose unimodal and multimodal denoising tasks to train MUCN with explicit supervision via a bi-level optimization strategy. We then jointly train unimodal and multimodal learning tasks to extract discriminative unimodal features for multimodal inference. Experimental results suggest that MUG outperforms competitive baselines and can learn accurate unimodal labels.
Relaxing Continuous Constraints of Equivariant Graph Neural Networks for Physical Dynamics Learning
Jun 24, 2024
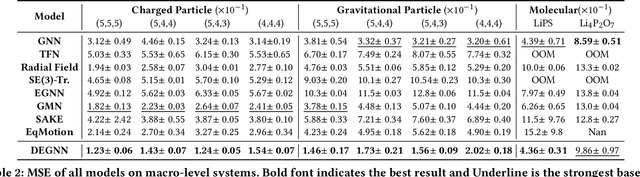


Incorporating Euclidean symmetries (e.g. rotation equivariance) as inductive biases into graph neural networks has improved their generalization ability and data efficiency in unbounded physical dynamics modeling. However, in various scientific and engineering applications, the symmetries of dynamics are frequently discrete due to the boundary conditions. Thus, existing GNNs either overlook necessary symmetry, resulting in suboptimal representation ability, or impose excessive equivariance, which fails to generalize to unobserved symmetric dynamics. In this work, we propose a general Discrete Equivariant Graph Neural Network (DEGNN) that guarantees equivariance to a given discrete point group. Specifically, we show that such discrete equivariant message passing could be constructed by transforming geometric features into permutation-invariant embeddings. Through relaxing continuous equivariant constraints, DEGNN can employ more geometric feature combinations to approximate unobserved physical object interaction functions. Two implementation approaches of DEGNN are proposed based on ranking or pooling permutation-invariant functions. We apply DEGNN to various physical dynamics, ranging from particle, molecular, crowd to vehicle dynamics. In twenty scenarios, DEGNN significantly outperforms existing state-of-the-art approaches. Moreover, we show that DEGNN is data efficient, learning with less data, and can generalize across scenarios such as unobserved orientation.
SciKnowEval: Evaluating Multi-level Scientific Knowledge of Large Language Models
Jun 13, 2024The burgeoning utilization of Large Language Models (LLMs) in scientific research necessitates advanced benchmarks capable of evaluating their understanding and application of scientific knowledge comprehensively. To address this need, we introduce the SciKnowEval benchmark, a novel framework that systematically evaluates LLMs across five progressive levels of scientific knowledge: studying extensively, inquiring earnestly, thinking profoundly, discerning clearly, and practicing assiduously. These levels aim to assess the breadth and depth of scientific knowledge in LLMs, including knowledge coverage, inquiry and exploration capabilities, reflection and reasoning abilities, ethic and safety considerations, as well as practice proficiency. Specifically, we take biology and chemistry as the two instances of SciKnowEval and construct a dataset encompassing 50K multi-level scientific problems and solutions. By leveraging this dataset, we benchmark 20 leading open-source and proprietary LLMs using zero-shot and few-shot prompting strategies. The results reveal that despite achieving state-of-the-art performance, the proprietary LLMs still have considerable room for improvement, particularly in addressing scientific computations and applications. We anticipate that SciKnowEval will establish a comprehensive standard for benchmarking LLMs in science research and discovery, and promote the development of LLMs that integrate scientific knowledge with strong safety awareness. The dataset and code are publicly available at https://github.com/hicai-zju/sciknoweval .
Atomas: Hierarchical Alignment on Molecule-Text for Unified Molecule Understanding and Generation
Apr 23, 2024Molecule-and-text cross-modal representation learning has emerged as a promising direction for enhancing the quality of molecular representation, thereby improving performance in various scientific fields, including drug discovery and materials science. Existing studies adopt a global alignment approach to learn the knowledge from different modalities. These global alignment approaches fail to capture fine-grained information, such as molecular fragments and their corresponding textual description, which is crucial for downstream tasks. Furthermore, it is incapable to model such information using a similar global alignment strategy due to data scarcity of paired local part annotated data from existing datasets. In this paper, we propose Atomas, a multi-modal molecular representation learning framework to jointly learn representations from SMILES string and text. We design a Hierarchical Adaptive Alignment model to concurrently learn the fine-grained fragment correspondence between two modalities and align these representations of fragments in three levels. Additionally, Atomas's end-to-end training framework incorporates the tasks of understanding and generating molecule, thereby supporting a wider range of downstream tasks. In the retrieval task, Atomas exhibits robust generalization ability and outperforms the baseline by 30.8% of recall@1 on average. In the generation task, Atomas achieves state-of-the-art results in both molecule captioning task and molecule generation task. Moreover, the visualization of the Hierarchical Adaptive Alignment model further confirms the chemical significance of our approach. Our codes can be found at https://anonymous.4open.science/r/Atomas-03C3.
Functional Protein Design with Local Domain Alignment
Apr 18, 2024



The core challenge of de novo protein design lies in creating proteins with specific functions or properties, guided by certain conditions. Current models explore to generate protein using structural and evolutionary guidance, which only provide indirect conditions concerning functions and properties. However, textual annotations of proteins, especially the annotations for protein domains, which directly describe the protein's high-level functionalities, properties, and their correlation with target amino acid sequences, remain unexplored in the context of protein design tasks. In this paper, we propose Protein-Annotation Alignment Generation (PAAG), a multi-modality protein design framework that integrates the textual annotations extracted from protein database for controllable generation in sequence space. Specifically, within a multi-level alignment module, PAAG can explicitly generate proteins containing specific domains conditioned on the corresponding domain annotations, and can even design novel proteins with flexible combinations of different kinds of annotations. Our experimental results underscore the superiority of the aligned protein representations from PAAG over 7 prediction tasks. Furthermore, PAAG demonstrates a nearly sixfold increase in generation success rate (24.7% vs 4.7% in zinc finger, and 54.3% vs 8.7% in the immunoglobulin domain) in comparison to the existing model.
StableMask: Refining Causal Masking in Decoder-only Transformer
Feb 07, 2024



The decoder-only Transformer architecture with causal masking and relative position encoding (RPE) has become the de facto choice in language modeling. Despite its exceptional performance across various tasks, we have identified two limitations: First, it requires all attention scores to be non-zero and sum up to 1, even if the current embedding has sufficient self-contained information. This compels the model to assign disproportional excessive attention to specific tokens. Second, RPE-based Transformers are not universal approximators due to their limited capacity at encoding absolute positional information, which limits their application in position-critical tasks. In this work, we propose StableMask: a parameter-free method to address both limitations by refining the causal mask. It introduces pseudo-attention values to balance attention distributions and encodes absolute positional information via a progressively decreasing mask ratio. StableMask's effectiveness is validated both theoretically and empirically, showing significant enhancements in language models with parameter sizes ranging from 71M to 1.4B across diverse datasets and encoding methods. We further show that it naturally supports (1) efficient extrapolation without special tricks such as StreamingLLM and (2) easy integration with existing attention optimization techniques.