 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeMed-Scout: Curing MLLMs' Geometric Blindness in Medical Perception via Geometry-Aware RL Post-Training
Jan 30, 2026Despite recent Multimodal Large Language Models (MLLMs)' linguistic prowess in medical diagnosis, we find even state-of-the-art MLLMs suffer from a critical perceptual deficit: geometric blindness. This failure to ground outputs in objective geometric constraints leads to plausible yet factually incorrect hallucinations, rooted in training paradigms that prioritize linguistic fluency over geometric fidelity. This paper introduces Med-Scout, a novel framework that "cures" this blindness via Reinforcement Learning (RL) that leverages the intrinsic geometric logic latent within unlabeled medical images. Instead of relying on costly expert annotations, Med-Scout derives verifiable supervision signals through three strategic proxy tasks: Hierarchical Scale Localization, Topological Jigsaw Reconstruction, and Anomaly Consistency Detection. To rigorously quantify this deficit, we present Med-Scout-Bench, a new benchmark specifically designed to evaluate geometric perception. Extensive evaluations show that Med-Scout significantly mitigates geometric blindness, outperforming leading proprietary and open-source MLLMs by over 40% on our benchmark. Furthermore, this enhanced geometric perception generalizes to broader medical understanding, achieving superior results on radiological and comprehensive medical VQA tasks.
ALEX:A Light Editing-knowledge Extractor
Nov 18, 2025


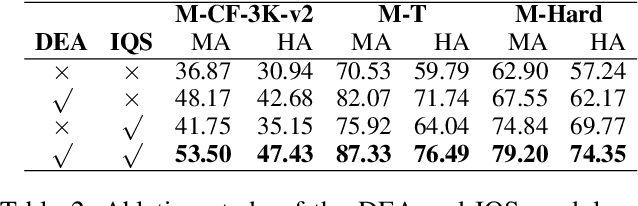
The static nature of knowledge within Large Language Models (LLMs) makes it difficult for them to adapt to evolving information, rendering knowledge editing a critical task. However, existing methods struggle with challenges of scalability and retrieval efficiency, particularly when handling complex, multi-hop questions that require multi-step reasoning. To address these challenges, this paper introduces ALEX (A Light Editing-knowledge Extractor), a lightweight knowledge editing framework. The core innovation of ALEX is its hierarchical memory architecture, which organizes knowledge updates (edits) into semantic clusters. This design fundamentally reduces retrieval complexity from a linear O(N) to a highly scalable O(K+N/C). Furthermore, the framework integrates an Inferential Query Synthesis (IQS) module to bridge the semantic gap between queries and facts , and a Dynamic Evidence Adjudication (DEA) engine that executes an efficient two-stage retrieval process. Experiments on the MQUAKE benchmark demonstrate that ALEX significantly improves both the accuracy of multi-hop answers (MultiHop-ACC) and the reliability of reasoning paths (HopWise-ACC). It also reduces the required search space by over 80% , presenting a promising path toward building scalable, efficient, and accurate knowledge editing systems.
WDT-MD: Wavelet Diffusion Transformers for Microaneurysm Detection in Fundus Images
Nov 16, 2025Microaneurysms (MAs), the earliest pathognomonic signs of Diabetic Retinopathy (DR), present as sub-60 $μm$ lesions in fundus images with highly variable photometric and morphological characteristics, rendering manual screening not only labor-intensive but inherently error-prone. While diffusion-based anomaly detection has emerged as a promising approach for automated MA screening, its clinical application is hindered by three fundamental limitations. First, these models often fall prey to "identity mapping", where they inadvertently replicate the input image. Second, they struggle to distinguish MAs from other anomalies, leading to high false positives. Third, their suboptimal reconstruction of normal features hampers overall performance. To address these challenges, we propose a Wavelet Diffusion Transformer framework for MA Detection (WDT-MD), which features three key innovations: a noise-encoded image conditioning mechanism to avoid "identity mapping" by perturbing image conditions during training; pseudo-normal pattern synthesis via inpainting to introduce pixel-level supervision, enabling discrimination between MAs and other anomalies; and a wavelet diffusion Transformer architecture that combines the global modeling capability of diffusion Transformers with multi-scale wavelet analysis to enhance reconstruction of normal retinal features. Comprehensive experiments on the IDRiD and e-ophtha MA datasets demonstrate that WDT-MD outperforms state-of-the-art methods in both pixel-level and image-level MA detection. This advancement holds significant promise for improving early DR screening.
Mitigating Hallucination of Large Vision-Language Models via Dynamic Logits Calibration
Jun 26, 2025Large Vision-Language Models (LVLMs) have demonstrated significant advancements in multimodal understanding, yet they are frequently hampered by hallucination-the generation of text that contradicts visual input. Existing training-free decoding strategies exhibit critical limitations, including the use of static constraints that do not adapt to semantic drift during generation, inefficiency stemming from the need for multiple forward passes, and degradation of detail due to overly rigid intervention rules. To overcome these challenges, this paper introduces Dynamic Logits Calibration (DLC), a novel training-free decoding framework designed to dynamically align text generation with visual evidence at inference time. At the decoding phase, DLC step-wise employs CLIP to assess the semantic alignment between the input image and the generated text sequence. Then, the Relative Visual Advantage (RVA) of candidate tokens is evaluated against a dynamically updated contextual baseline, adaptively adjusting output logits to favor tokens that are visually grounded. Furthermore, an adaptive weighting mechanism, informed by a real-time context alignment score, carefully balances the visual guidance while ensuring the overall quality of the textual output. Extensive experiments conducted across diverse benchmarks and various LVLM architectures (such as LLaVA, InstructBLIP, and MiniGPT-4) demonstrate that DLC significantly reduces hallucinations, outperforming current methods while maintaining high inference efficiency by avoiding multiple forward passes. Overall, we present an effective and efficient decoding-time solution to mitigate hallucinations, thereby enhancing the reliability of LVLMs for more practices. Code will be released on Github.
Med-U1: Incentivizing Unified Medical Reasoning in LLMs via Large-scale Reinforcement Learning
Jun 14, 2025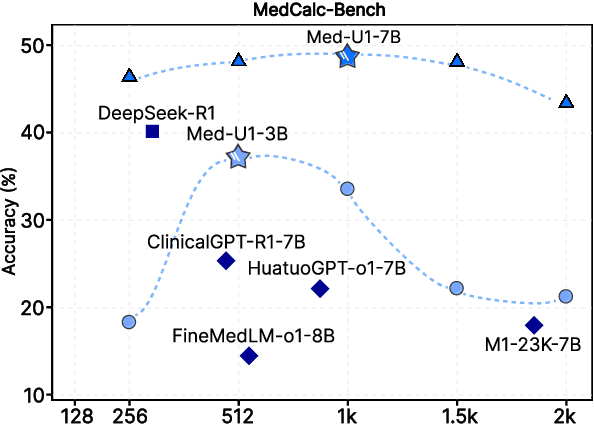
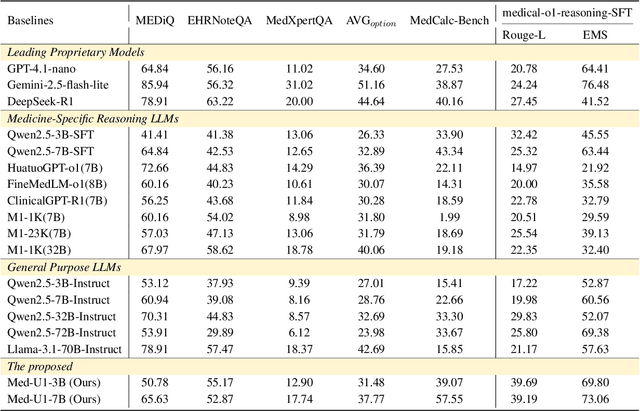
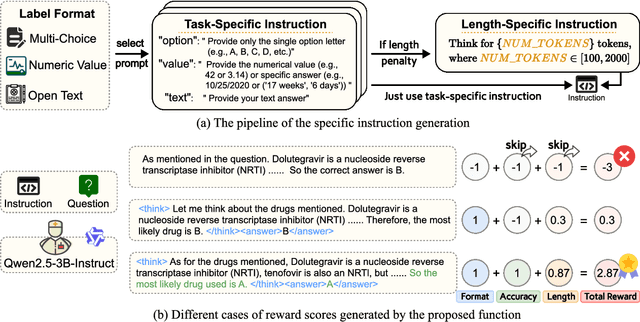
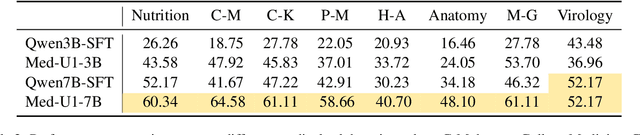
Medical Question-Answering (QA) encompasses a broad spectrum of tasks, including multiple choice questions (MCQ), open-ended text generation, and complex computational reasoning. Despite this variety, a unified framework for delivering high-quality medical QA has yet to emerge. Although recent progress in reasoning-augmented large language models (LLMs) has shown promise, their ability to achieve comprehensive medical understanding is still largely unexplored. In this paper, we present Med-U1, a unified framework for robust reasoning across medical QA tasks with diverse output formats, ranging from MCQs to complex generation and computation tasks. Med-U1 employs pure large-scale reinforcement learning with mixed rule-based binary reward functions, incorporating a length penalty to manage output verbosity. With multi-objective reward optimization, Med-U1 directs LLMs to produce concise and verifiable reasoning chains. Empirical results reveal that Med-U1 significantly improves performance across multiple challenging Med-QA benchmarks, surpassing even larger specialized and proprietary models. Furthermore, Med-U1 demonstrates robust generalization to out-of-distribution (OOD) tasks. Extensive analysis presents insights into training strategies, reasoning chain length control, and reward design for medical LLMs. The code will be released.
Uncertainty-Aware Multi-Expert Knowledge Distillation for Imbalanced Disease Grading
May 01, 2025Automatic disease image grading is a significant application of artificial intelligence for healthcare, enabling faster and more accurate patient assessments. However, domain shifts, which are exacerbated by data imbalance, introduce bias into the model, posing deployment difficulties in clinical applications. To address the problem, we propose a novel \textbf{U}ncertainty-aware \textbf{M}ulti-experts \textbf{K}nowledge \textbf{D}istillation (UMKD) framework to transfer knowledge from multiple expert models to a single student model. Specifically, to extract discriminative features, UMKD decouples task-agnostic and task-specific features with shallow and compact feature alignment in the feature space. At the output space, an uncertainty-aware decoupled distillation (UDD) mechanism dynamically adjusts knowledge transfer weights based on expert model uncertainties, ensuring robust and reliable distillation. Additionally, UMKD also tackles the problems of model architecture heterogeneity and distribution discrepancies between source and target domains, which are inadequately tackled by previous KD approaches. Extensive experiments on histology prostate grading (\textit{SICAPv2}) and fundus image grading (\textit{APTOS}) demonstrate that UMKD achieves a new state-of-the-art in both source-imbalanced and target-imbalanced scenarios, offering a robust and practical solution for real-world disease image grading.
Matrix Factorization with Dynamic Multi-view Clustering for Recommender System
Apr 20, 2025Matrix factorization (MF), a cornerstone of recommender systems, decomposes user-item interaction matrices into latent representations. Traditional MF approaches, however, employ a two-stage, non-end-to-end paradigm, sequentially performing recommendation and clustering, resulting in prohibitive computational costs for large-scale applications like e-commerce and IoT, where billions of users interact with trillions of items. To address this, we propose Matrix Factorization with Dynamic Multi-view Clustering (MFDMC), a unified framework that balances efficient end-to-end training with comprehensive utilization of web-scale data and enhances interpretability. MFDMC leverages dynamic multi-view clustering to learn user and item representations, adaptively pruning poorly formed clusters. Each entity's representation is modeled as a weighted projection of robust clusters, capturing its diverse roles across views. This design maximizes representation space utilization, improves interpretability, and ensures resilience for downstream tasks. Extensive experiments demonstrate MFDMC's superior performance in recommender systems and other representation learning domains, such as computer vision, highlighting its scalability and versatility.
ProtFlow: Fast Protein Sequence Design via Flow Matching on Compressed Protein Language Model Embeddings
Apr 15, 2025The design of protein sequences with desired functionalities is a fundamental task in protein engineering. Deep generative methods, such as autoregressive models and diffusion models, have greatly accelerated the discovery of novel protein sequences. However, these methods mainly focus on local or shallow residual semantics and suffer from low inference efficiency, large modeling space and high training cost. To address these challenges, we introduce ProtFlow, a fast flow matching-based protein sequence design framework that operates on embeddings derived from semantically meaningful latent space of protein language models. By compressing and smoothing the latent space, ProtFlow enhances performance while training on limited computational resources. Leveraging reflow techniques, ProtFlow enables high-quality single-step sequence generation. Additionally, we develop a joint design pipeline for the design scene of multichain proteins. We evaluate ProtFlow across diverse protein design tasks, including general peptides and long-chain proteins, antimicrobial peptides, and antibodies. Experimental results demonstrate that ProtFlow outperforms task-specific methods in these applications, underscoring its potential and broad applicability in computational protein sequence design and analysis.
From Misleading Queries to Accurate Answers: A Three-Stage Fine-Tuning Method for LLMs
Apr 15, 2025



Large language models (LLMs) exhibit excellent performance in natural language processing (NLP), but remain highly sensitive to the quality of input queries, especially when these queries contain misleading or inaccurate information. Existing methods focus on correcting the output, but they often overlook the potential of improving the ability of LLMs to detect and correct misleading content in the input itself. In this paper, we propose a novel three-stage fine-tuning method that enhances the ability of LLMs to detect and correct misleading information in the input, further improving response accuracy and reducing hallucinations. Specifically, the three stages include (1) training LLMs to identify misleading information, (2) training LLMs to correct the misleading information using built-in or external knowledge, and (3) training LLMs to generate accurate answers based on the corrected queries. To evaluate our method, we conducted experiments on three datasets for the hallucination detection task and the question answering (QA) task, as well as two datasets containing misleading information that we constructed. The experimental results demonstrate that our method significantly improves the accuracy and factuality of LLM responses, while also enhancing the ability to detect hallucinations and reducing the generation of hallucinations in the output, particularly when the query contains misleading information. We will publicly release our code upon acceptance.
OrderChain: A General Prompting Paradigm to Improve Ordinal Understanding Ability of MLLM
Apr 07, 2025Despite the remarkable progress of multimodal large language models (MLLMs), they continue to face challenges in achieving competitive performance on ordinal regression (OR; a.k.a. ordinal classification). To address this issue, this paper presents OrderChain, a novel and general prompting paradigm that improves the ordinal understanding ability of MLLMs by specificity and commonality modeling. Specifically, our OrderChain consists of a set of task-aware prompts to facilitate the specificity modeling of diverse OR tasks and a new range optimization Chain-of-Thought (RO-CoT), which learns a commonality way of thinking about OR tasks by uniformly decomposing them into multiple small-range optimization subtasks. Further, we propose a category recursive division (CRD) method to generate instruction candidate category prompts to support RO-CoT automatic optimization. Comprehensive experiments show that a Large Language and Vision Assistant (LLaVA) model with our OrderChain improves baseline LLaVA significantly on diverse OR datasets, e.g., from 47.5% to 93.2% accuracy on the Adience dataset for age estimation, and from 30.0% to 85.7% accuracy on the Diabetic Retinopathy dataset. Notably, LLaVA with our OrderChain also remarkably outperforms state-of-the-art methods by 27% on accuracy and 0.24 on MAE on the Adience dataset. To our best knowledge, our OrderChain is the first work that augments MLLMs for OR tasks, and the effectiveness is witnessed across a spectrum of OR datasets.