 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeProtFlow: Fast Protein Sequence Design via Flow Matching on Compressed Protein Language Model Embeddings
Apr 15, 2025The design of protein sequences with desired functionalities is a fundamental task in protein engineering. Deep generative methods, such as autoregressive models and diffusion models, have greatly accelerated the discovery of novel protein sequences. However, these methods mainly focus on local or shallow residual semantics and suffer from low inference efficiency, large modeling space and high training cost. To address these challenges, we introduce ProtFlow, a fast flow matching-based protein sequence design framework that operates on embeddings derived from semantically meaningful latent space of protein language models. By compressing and smoothing the latent space, ProtFlow enhances performance while training on limited computational resources. Leveraging reflow techniques, ProtFlow enables high-quality single-step sequence generation. Additionally, we develop a joint design pipeline for the design scene of multichain proteins. We evaluate ProtFlow across diverse protein design tasks, including general peptides and long-chain proteins, antimicrobial peptides, and antibodies. Experimental results demonstrate that ProtFlow outperforms task-specific methods in these applications, underscoring its potential and broad applicability in computational protein sequence design and analysis.
Efficient Autoregressive Shape Generation via Octree-Based Adaptive Tokenization
Apr 03, 2025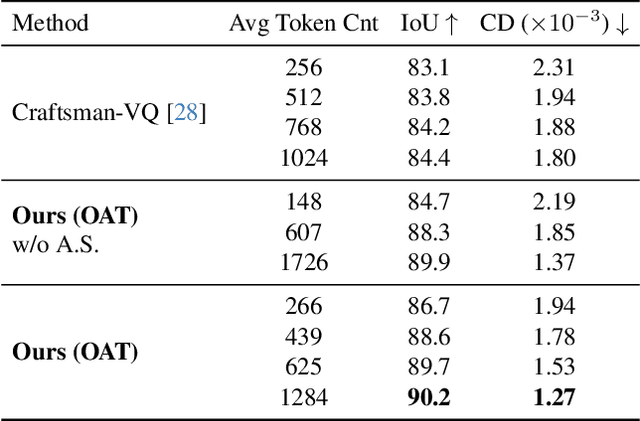

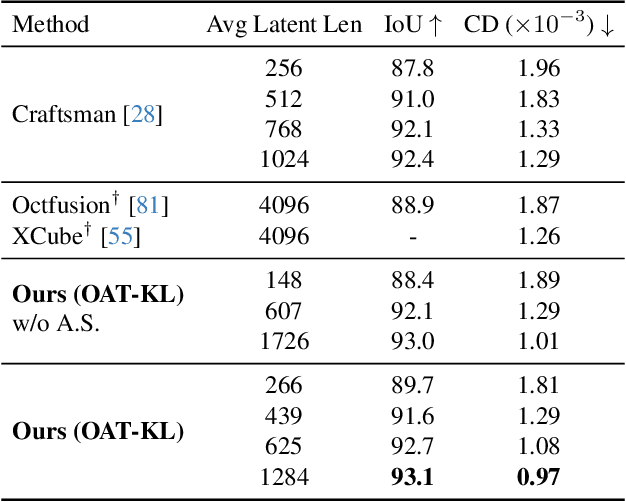
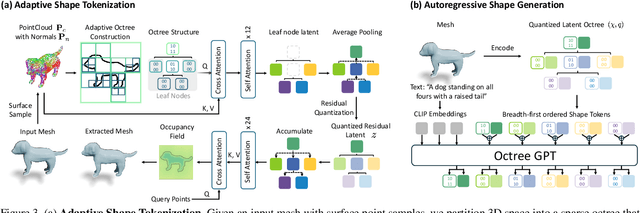
Many 3D generative models rely on variational autoencoders (VAEs) to learn compact shape representations. However, existing methods encode all shapes into a fixed-size token, disregarding the inherent variations in scale and complexity across 3D data. This leads to inefficient latent representations that can compromise downstream generation. We address this challenge by introducing Octree-based Adaptive Tokenization, a novel framework that adjusts the dimension of latent representations according to shape complexity. Our approach constructs an adaptive octree structure guided by a quadric-error-based subdivision criterion and allocates a shape latent vector to each octree cell using a query-based transformer. Building upon this tokenization, we develop an octree-based autoregressive generative model that effectively leverages these variable-sized representations in shape generation. Extensive experiments demonstrate that our approach reduces token counts by 50% compared to fixed-size methods while maintaining comparable visual quality. When using a similar token length, our method produces significantly higher-quality shapes. When incorporated with our downstream generative model, our method creates more detailed and diverse 3D content than existing approaches.
Cube: A Roblox View of 3D Intelligence
Mar 19, 2025



Foundation models trained on vast amounts of data have demonstrated remarkable reasoning and generation capabilities in the domains of text, images, audio and video. Our goal at Roblox is to build such a foundation model for 3D intelligence, a model that can support developers in producing all aspects of a Roblox experience, from generating 3D objects and scenes to rigging characters for animation to producing programmatic scripts describing object behaviors. We discuss three key design requirements for such a 3D foundation model and then present our first step towards building such a model. We expect that 3D geometric shapes will be a core data type and describe our solution for 3D shape tokenizer. We show how our tokenization scheme can be used in applications for text-to-shape generation, shape-to-text generation and text-to-scene generation. We demonstrate how these applications can collaborate with existing large language models (LLMs) to perform scene analysis and reasoning. We conclude with a discussion outlining our path to building a fully unified foundation model for 3D intelligence.
A Generalist Cross-Domain Molecular Learning Framework for Structure-Based Drug Discovery
Mar 06, 2025Structure-based drug discovery (SBDD) is a systematic scientific process that develops new drugs by leveraging the detailed physical structure of the target protein. Recent advancements in pre-trained models for biomolecules have demonstrated remarkable success across various biochemical applications, including drug discovery and protein engineering. However, in most approaches, the pre-trained models primarily focus on the characteristics of either small molecules or proteins, without delving into their binding interactions which are essential cross-domain relationships pivotal to SBDD. To fill this gap, we propose a general-purpose foundation model named BIT (an abbreviation for Biomolecular Interaction Transformer), which is capable of encoding a range of biochemical entities, including small molecules, proteins, and protein-ligand complexes, as well as various data formats, encompassing both 2D and 3D structures. Specifically, we introduce Mixture-of-Domain-Experts (MoDE) to handle the biomolecules from diverse biochemical domains and Mixture-of-Structure-Experts (MoSE) to capture positional dependencies in the molecular structures. The proposed mixture-of-experts approach enables BIT to achieve both deep fusion and domain-specific encoding, effectively capturing fine-grained molecular interactions within protein-ligand complexes. Then, we perform cross-domain pre-training on the shared Transformer backbone via several unified self-supervised denoising tasks. Experimental results on various benchmarks demonstrate that BIT achieves exceptional performance in downstream tasks, including binding affinity prediction, structure-based virtual screening, and molecular property prediction.
S$^2$ALM: Sequence-Structure Pre-trained Large Language Model for Comprehensive Antibody Representation Learning
Nov 20, 2024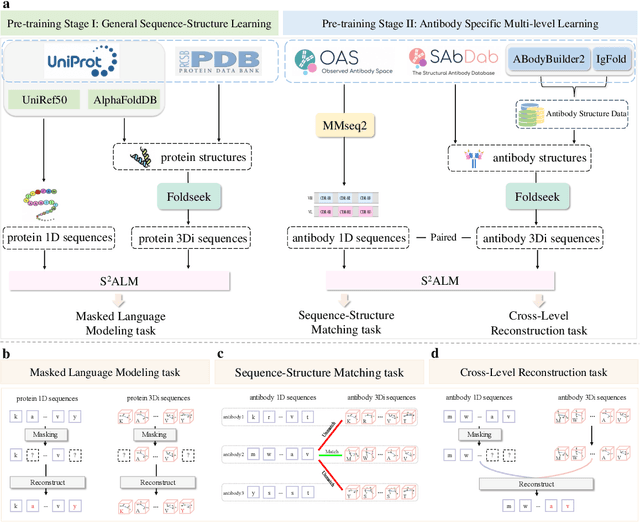
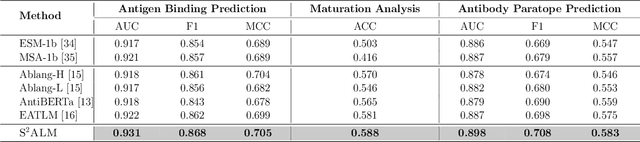
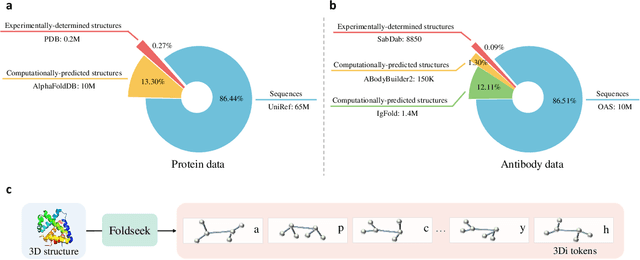
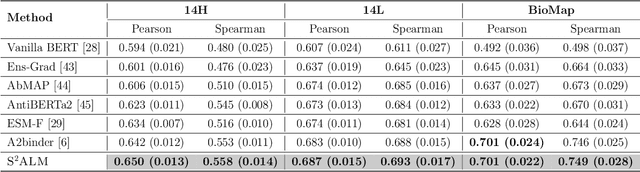
Antibodies safeguard our health through their precise and potent binding to specific antigens, demonstrating promising therapeutic efficacy in the treatment of numerous diseases, including COVID-19. Recent advancements in biomedical language models have shown the great potential to interpret complex biological structures and functions. However, existing antibody specific models have a notable limitation that they lack explicit consideration for antibody structural information, despite the fact that both 1D sequence and 3D structure carry unique and complementary insights into antibody behavior and functionality. This paper proposes Sequence-Structure multi-level pre-trained Antibody Language Model (S$^2$ALM), combining holistic sequential and structural information in one unified, generic antibody foundation model. We construct a hierarchical pre-training paradigm incorporated with two customized multi-level training objectives to facilitate the modeling of comprehensive antibody representations. S$^2$ALM's representation space uncovers inherent functional binding mechanisms, biological evolution properties and structural interaction patterns. Pre-trained over 75 million sequences and 11.7 million structures, S$^2$ALM can be adopted for diverse downstream tasks: accurately predicting antigen-antibody binding affinities, precisely distinguishing B cell maturation stages, identifying antibody crucial binding positions, and specifically designing novel coronavirus-binding antibodies. Remarkably, S$^2$ALM outperforms well-established and renowned baselines and sets new state-of-the-art performance across extensive antibody specific understanding and generation tasks. S$^2$ALM's ability to model comprehensive and generalized representations further positions its potential to advance real-world therapeutic antibody development, potentially addressing unmet academic, industrial, and clinical needs.
Bridge-IF: Learning Inverse Protein Folding with Markov Bridges
Nov 04, 2024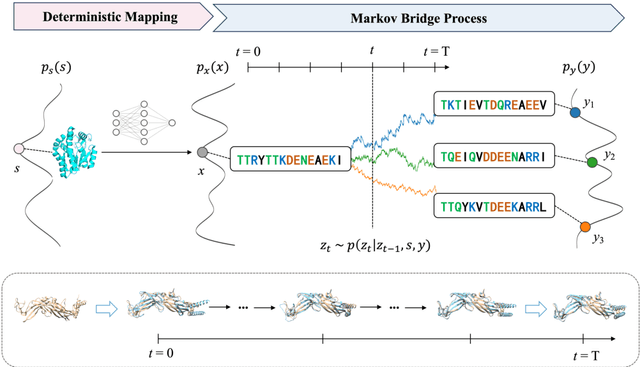
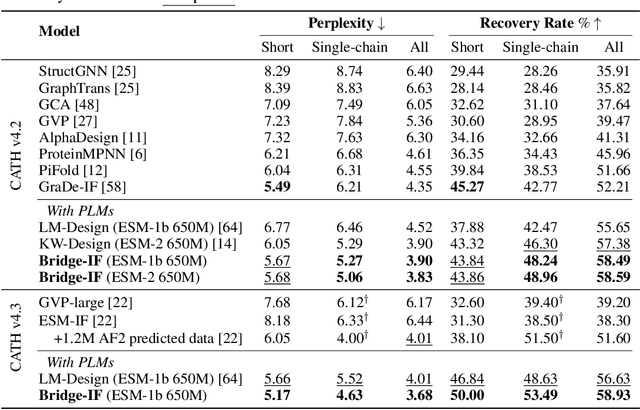
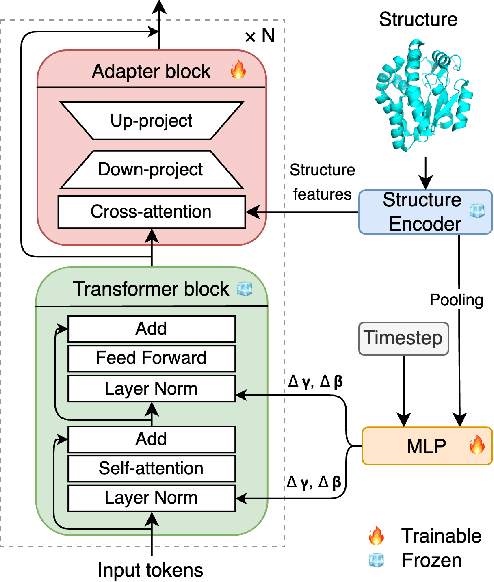
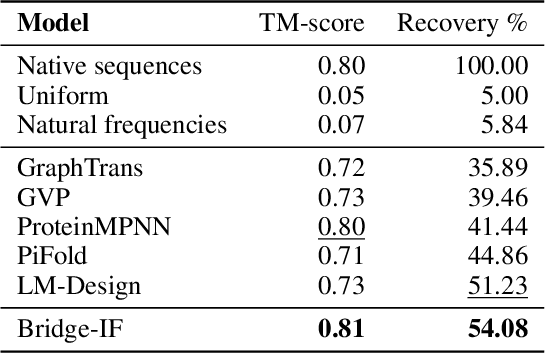
Inverse protein folding is a fundamental task in computational protein design, which aims to design protein sequences that fold into the desired backbone structures. While the development of machine learning algorithms for this task has seen significant success, the prevailing approaches, which predominantly employ a discriminative formulation, frequently encounter the error accumulation issue and often fail to capture the extensive variety of plausible sequences. To fill these gaps, we propose Bridge-IF, a generative diffusion bridge model for inverse folding, which is designed to learn the probabilistic dependency between the distributions of backbone structures and protein sequences. Specifically, we harness an expressive structure encoder to propose a discrete, informative prior derived from structures, and establish a Markov bridge to connect this prior with native sequences. During the inference stage, Bridge-IF progressively refines the prior sequence, culminating in a more plausible design. Moreover, we introduce a reparameterization perspective on Markov bridge models, from which we derive a simplified loss function that facilitates more effective training. We also modulate protein language models (PLMs) with structural conditions to precisely approximate the Markov bridge process, thereby significantly enhancing generation performance while maintaining parameter-efficient training. Extensive experiments on well-established benchmarks demonstrate that Bridge-IF predominantly surpasses existing baselines in sequence recovery and excels in the design of plausible proteins with high foldability. The code is available at https://github.com/violet-sto/Bridge-IF.
Structure-Enhanced Protein Instruction Tuning: Towards General-Purpose Protein Understanding
Oct 04, 2024Proteins, as essential biomolecules, play a central role in biological processes, including metabolic reactions and DNA replication. Accurate prediction of their properties and functions is crucial in biological applications. Recent development of protein language models (pLMs) with supervised fine tuning provides a promising solution to this problem. However, the fine-tuned model is tailored for particular downstream prediction task, and achieving general-purpose protein understanding remains a challenge. In this paper, we introduce Structure-Enhanced Protein Instruction Tuning (SEPIT) framework to bridge this gap. Our approach integrates a noval structure-aware module into pLMs to inform them with structural knowledge, and then connects these enhanced pLMs to large language models (LLMs) to generate understanding of proteins. In this framework, we propose a novel two-stage instruction tuning pipeline that first establishes a basic understanding of proteins through caption-based instructions and then refines this understanding using a mixture of experts (MoEs) to learn more complex properties and functional information with the same amount of activated parameters. Moreover, we construct the largest and most comprehensive protein instruction dataset to date, which allows us to train and evaluate the general-purpose protein understanding model. Extensive experimental results on open-ended generation and closed-set answer tasks demonstrate the superior performance of SEPIT over both closed-source general LLMs and open-source LLMs trained with protein knowledge.
Multi-Modal CLIP-Informed Protein Editing
Jul 27, 2024Proteins govern most biological functions essential for life, but achieving controllable protein discovery and optimization remains challenging. Recently, machine learning-assisted protein editing (MLPE) has shown promise in accelerating optimization cycles and reducing experimental workloads. However, current methods struggle with the vast combinatorial space of potential protein edits and cannot explicitly conduct protein editing using biotext instructions, limiting their interactivity with human feedback. To fill these gaps, we propose a novel method called ProtET for efficient CLIP-informed protein editing through multi-modality learning. Our approach comprises two stages: in the pretraining stage, contrastive learning aligns protein-biotext representations encoded by two large language models (LLMs), respectively. Subsequently, during the protein editing stage, the fused features from editing instruction texts and original protein sequences serve as the final editing condition for generating target protein sequences. Comprehensive experiments demonstrated the superiority of ProtET in editing proteins to enhance human-expected functionality across multiple attribute domains, including enzyme catalytic activity, protein stability and antibody specific binding ability. And ProtET improves the state-of-the-art results by a large margin, leading to significant stability improvements of 16.67% and 16.90%. This capability positions ProtET to advance real-world artificial protein editing, potentially addressing unmet academic, industrial, and clinical needs.
Dr3: Ask Large Language Models Not to Give Off-Topic Answers in Open Domain Multi-Hop Question Answering
Mar 19, 2024



Open Domain Multi-Hop Question Answering (ODMHQA) plays a crucial role in Natural Language Processing (NLP) by aiming to answer complex questions through multi-step reasoning over retrieved information from external knowledge sources. Recently, Large Language Models (LLMs) have demonstrated remarkable performance in solving ODMHQA owing to their capabilities including planning, reasoning, and utilizing tools. However, LLMs may generate off-topic answers when attempting to solve ODMHQA, namely the generated answers are irrelevant to the original questions. This issue of off-topic answers accounts for approximately one-third of incorrect answers, yet remains underexplored despite its significance. To alleviate this issue, we propose the Discriminate->Re-Compose->Re- Solve->Re-Decompose (Dr3) mechanism. Specifically, the Discriminator leverages the intrinsic capabilities of LLMs to judge whether the generated answers are off-topic. In cases where an off-topic answer is detected, the Corrector performs step-wise revisions along the reversed reasoning chain (Re-Compose->Re-Solve->Re-Decompose) until the final answer becomes on-topic. Experimental results on the HotpotQA and 2WikiMultiHopQA datasets demonstrate that our Dr3 mechanism considerably reduces the occurrence of off-topic answers in ODMHQA by nearly 13%, improving the performance in Exact Match (EM) by nearly 3% compared to the baseline method without the Dr3 mechanism.
Making Pre-trained Language Models Great on Tabular Prediction
Mar 12, 2024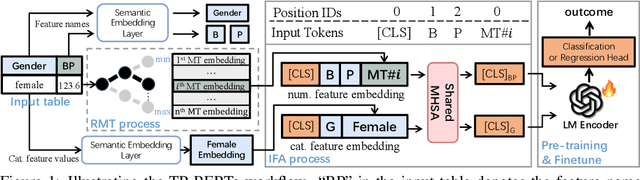

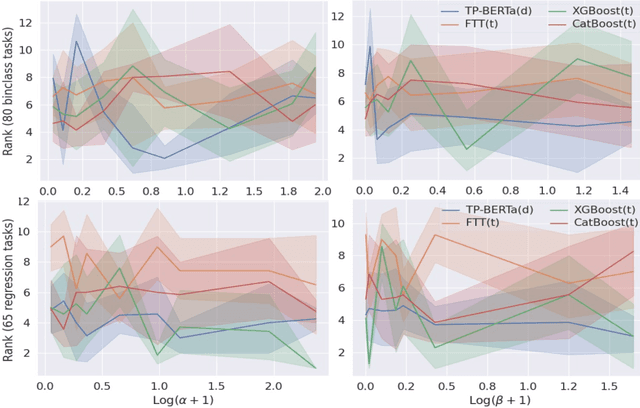

The transferability of deep neural networks (DNNs) has made significant progress in image and language processing. However, due to the heterogeneity among tables, such DNN bonus is still far from being well exploited on tabular data prediction (e.g., regression or classification tasks). Condensing knowledge from diverse domains, language models (LMs) possess the capability to comprehend feature names from various tables, potentially serving as versatile learners in transferring knowledge across distinct tables and diverse prediction tasks, but their discrete text representation space is inherently incompatible with numerical feature values in tables. In this paper, we present TP-BERTa, a specifically pre-trained LM for tabular data prediction. Concretely, a novel relative magnitude tokenization converts scalar numerical feature values to finely discrete, high-dimensional tokens, and an intra-feature attention approach integrates feature values with the corresponding feature names. Comprehensive experiments demonstrate that our pre-trained TP-BERTa leads the performance among tabular DNNs and is competitive with Gradient Boosted Decision Tree models in typical tabular data regime.