 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeA Generalist Cross-Domain Molecular Learning Framework for Structure-Based Drug Discovery
Mar 06, 2025Structure-based drug discovery (SBDD) is a systematic scientific process that develops new drugs by leveraging the detailed physical structure of the target protein. Recent advancements in pre-trained models for biomolecules have demonstrated remarkable success across various biochemical applications, including drug discovery and protein engineering. However, in most approaches, the pre-trained models primarily focus on the characteristics of either small molecules or proteins, without delving into their binding interactions which are essential cross-domain relationships pivotal to SBDD. To fill this gap, we propose a general-purpose foundation model named BIT (an abbreviation for Biomolecular Interaction Transformer), which is capable of encoding a range of biochemical entities, including small molecules, proteins, and protein-ligand complexes, as well as various data formats, encompassing both 2D and 3D structures. Specifically, we introduce Mixture-of-Domain-Experts (MoDE) to handle the biomolecules from diverse biochemical domains and Mixture-of-Structure-Experts (MoSE) to capture positional dependencies in the molecular structures. The proposed mixture-of-experts approach enables BIT to achieve both deep fusion and domain-specific encoding, effectively capturing fine-grained molecular interactions within protein-ligand complexes. Then, we perform cross-domain pre-training on the shared Transformer backbone via several unified self-supervised denoising tasks. Experimental results on various benchmarks demonstrate that BIT achieves exceptional performance in downstream tasks, including binding affinity prediction, structure-based virtual screening, and molecular property prediction.
GENERator: A Long-Context Generative Genomic Foundation Model
Feb 11, 2025Advancements in DNA sequencing technologies have significantly improved our ability to decode genomic sequences. However, the prediction and interpretation of these sequences remain challenging due to the intricate nature of genetic material. Large language models (LLMs) have introduced new opportunities for biological sequence analysis. Recent developments in genomic language models have underscored the potential of LLMs in deciphering DNA sequences. Nonetheless, existing models often face limitations in robustness and application scope, primarily due to constraints in model structure and training data scale. To address these limitations, we present GENERator, a generative genomic foundation model featuring a context length of 98k base pairs (bp) and 1.2B parameters. Trained on an expansive dataset comprising 386B bp of eukaryotic DNA, the GENERator demonstrates state-of-the-art performance across both established and newly proposed benchmarks. The model adheres to the central dogma of molecular biology, accurately generating protein-coding sequences that translate into proteins structurally analogous to known families. It also shows significant promise in sequence optimization, particularly through the prompt-responsive generation of promoter sequences with specific activity profiles. These capabilities position the GENERator as a pivotal tool for genomic research and biotechnological advancement, enhancing our ability to interpret and predict complex biological systems and enabling precise genomic interventions.
Bridge-IF: Learning Inverse Protein Folding with Markov Bridges
Nov 04, 2024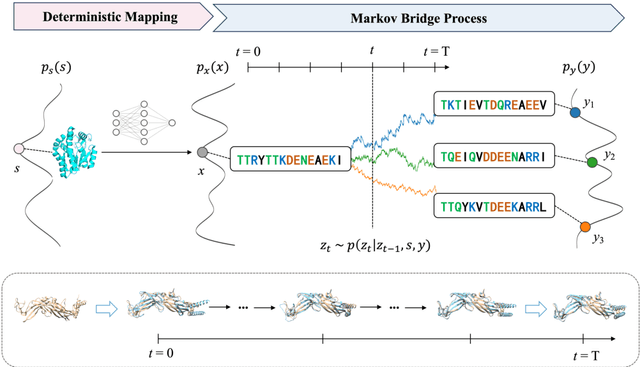
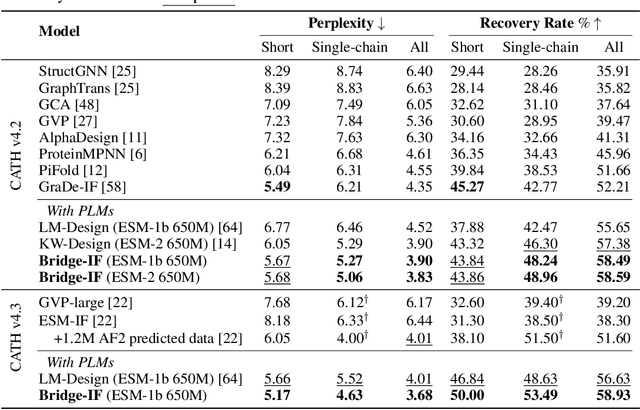
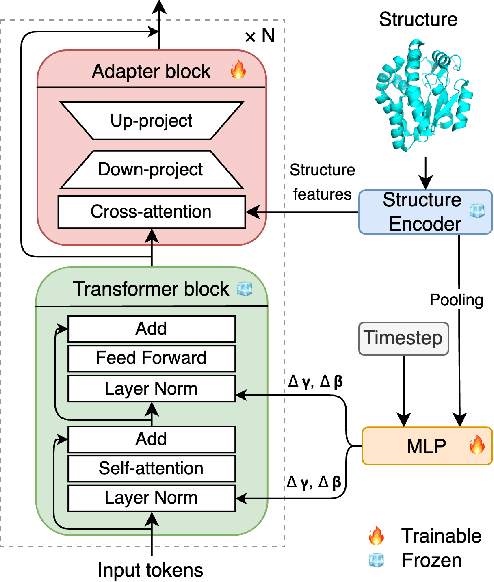
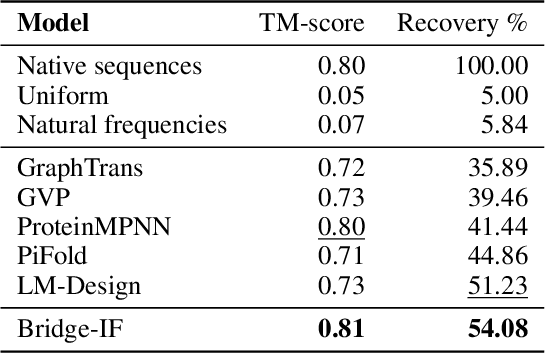
Inverse protein folding is a fundamental task in computational protein design, which aims to design protein sequences that fold into the desired backbone structures. While the development of machine learning algorithms for this task has seen significant success, the prevailing approaches, which predominantly employ a discriminative formulation, frequently encounter the error accumulation issue and often fail to capture the extensive variety of plausible sequences. To fill these gaps, we propose Bridge-IF, a generative diffusion bridge model for inverse folding, which is designed to learn the probabilistic dependency between the distributions of backbone structures and protein sequences. Specifically, we harness an expressive structure encoder to propose a discrete, informative prior derived from structures, and establish a Markov bridge to connect this prior with native sequences. During the inference stage, Bridge-IF progressively refines the prior sequence, culminating in a more plausible design. Moreover, we introduce a reparameterization perspective on Markov bridge models, from which we derive a simplified loss function that facilitates more effective training. We also modulate protein language models (PLMs) with structural conditions to precisely approximate the Markov bridge process, thereby significantly enhancing generation performance while maintaining parameter-efficient training. Extensive experiments on well-established benchmarks demonstrate that Bridge-IF predominantly surpasses existing baselines in sequence recovery and excels in the design of plausible proteins with high foldability. The code is available at https://github.com/violet-sto/Bridge-IF.