 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeVectorized Adaptive Histograms for Sparse Oblique Forests
Feb 27, 2026Classification using sparse oblique random forests provides guarantees on uncertainty and confidence while controlling for specific error types. However, they use more data and more compute than other tree ensembles because they create deep trees and need to sort or histogram linear combinations of data at runtime. We provide a method for dynamically switching between histograms and sorting to find the best split. We further optimize histogram construction using vector intrinsics. Evaluating this on large datasets, our optimizations speedup training by 1.7-2.5x compared to existing oblique forests and 1.5-2x compared to standard random forests. We also provide a GPU and hybrid CPU-GPU implementation.
Accelerating MHC-II Epitope Discovery via Multi-Scale Prediction in Antigen Presentation
Dec 16, 2025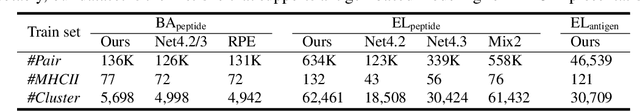
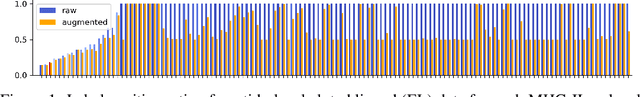
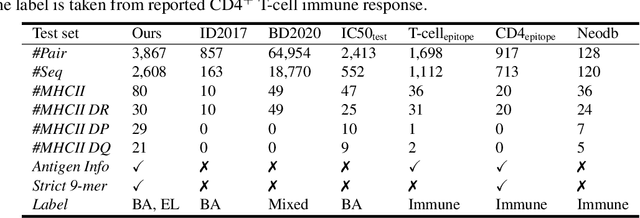
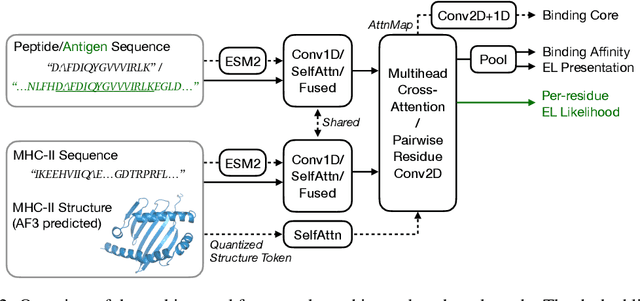
Antigenic epitope presented by major histocompatibility complex II (MHC-II) proteins plays an essential role in immunotherapy. However, compared to the more widely studied MHC-I in computational immunotherapy, the study of MHC-II antigenic epitope poses significantly more challenges due to its complex binding specificity and ambiguous motif patterns. Consequently, existing datasets for MHC-II interactions are smaller and less standardized than those available for MHC-I. To address these challenges, we present a well-curated dataset derived from the Immune Epitope Database (IEDB) and other public sources. It not only extends and standardizes existing peptide-MHC-II datasets, but also introduces a novel antigen-MHC-II dataset with richer biological context. Leveraging this dataset, we formulate three major machine learning (ML) tasks of peptide binding, peptide presentation, and antigen presentation, which progressively capture the broader biological processes within the MHC-II antigen presentation pathway. We further employ a multi-scale evaluation framework to benchmark existing models, along with a comprehensive analysis over various modeling designs to this problem with a modular framework. Overall, this work serves as a valuable resource for advancing computational immunotherapy, providing a foundation for future research in ML guided epitope discovery and predictive modeling of immune responses.
Unveiling Confirmation Bias in Chain-of-Thought Reasoning
Jun 14, 2025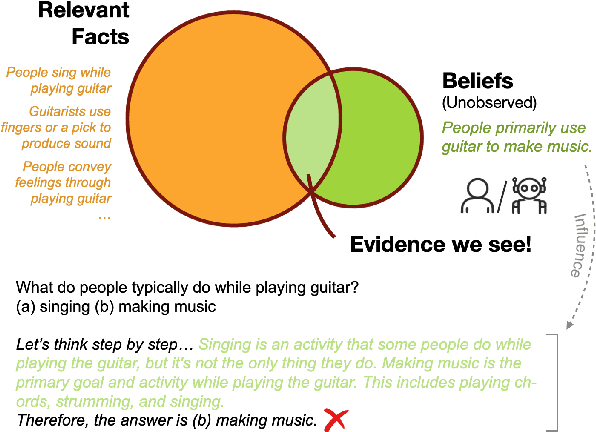
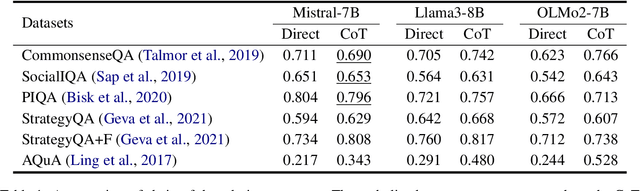
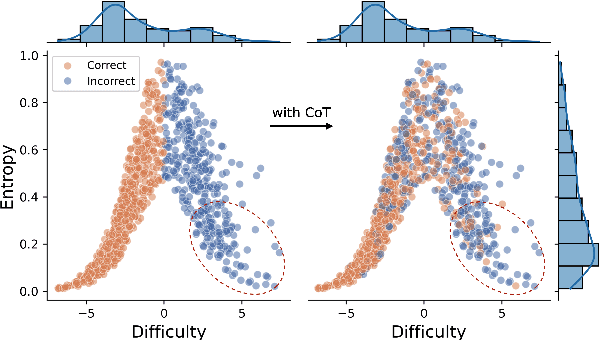
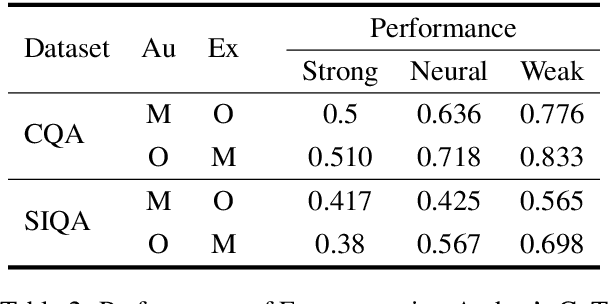
Chain-of-thought (CoT) prompting has been widely adopted to enhance the reasoning capabilities of large language models (LLMs). However, the effectiveness of CoT reasoning is inconsistent across tasks with different reasoning types. This work presents a novel perspective to understand CoT behavior through the lens of \textit{confirmation bias} in cognitive psychology. Specifically, we examine how model internal beliefs, approximated by direct question-answering probabilities, affect both reasoning generation ($Q \to R$) and reasoning-guided answer prediction ($QR \to A$) in CoT. By decomposing CoT into a two-stage process, we conduct a thorough correlation analysis in model beliefs, rationale attributes, and stage-wise performance. Our results provide strong evidence of confirmation bias in LLMs, such that model beliefs not only skew the reasoning process but also influence how rationales are utilized for answer prediction. Furthermore, the interplay between task vulnerability to confirmation bias and the strength of beliefs also provides explanations for CoT effectiveness across reasoning tasks and models. Overall, this study provides a valuable insight for the needs of better prompting strategies that mitigate confirmation bias to enhance reasoning performance. Code is available at \textit{https://github.com/yuewan2/biasedcot}.
RiNALMo: General-Purpose RNA Language Models Can Generalize Well on Structure Prediction Tasks
Feb 29, 2024Ribonucleic acid (RNA) plays a variety of crucial roles in fundamental biological processes. Recently, RNA has become an interesting drug target, emphasizing the need to improve our understanding of its structures and functions. Over the years, sequencing technologies have produced an enormous amount of unlabeled RNA data, which hides important knowledge and potential. Motivated by the successes of protein language models, we introduce RiboNucleic Acid Language Model (RiNALMo) to help unveil the hidden code of RNA. RiNALMo is the largest RNA language model to date with $650$ million parameters pre-trained on $36$ million non-coding RNA sequences from several available databases. RiNALMo is able to extract hidden knowledge and capture the underlying structure information implicitly embedded within the RNA sequences. RiNALMo achieves state-of-the-art results on several downstream tasks. Notably, we show that its generalization capabilities can overcome the inability of other deep learning methods for secondary structure prediction to generalize on unseen RNA families. The code has been made publicly available on https://github.com/lbcb-sci/RiNALMo.
Improving Explainable Object-induced Model through Uncertainty for Automated Vehicles
Feb 23, 2024



The rapid evolution of automated vehicles (AVs) has the potential to provide safer, more efficient, and comfortable travel options. However, these systems face challenges regarding reliability in complex driving scenarios. Recent explainable AV architectures neglect crucial information related to inherent uncertainties while providing explanations for actions. To overcome such challenges, our study builds upon the "object-induced" model approach that prioritizes the role of objects in scenes for decision-making and integrates uncertainty assessment into the decision-making process using an evidential deep learning paradigm with a Beta prior. Additionally, we explore several advanced training strategies guided by uncertainty, including uncertainty-guided data reweighting and augmentation. Leveraging the BDD-OIA dataset, our findings underscore that the model, through these enhancements, not only offers a clearer comprehension of AV decisions and their underlying reasoning but also surpasses existing baselines across a broad range of scenarios.
From molecules to scaffolds to functional groups: building context-dependent molecular representation via multi-channel learning
Nov 05, 2023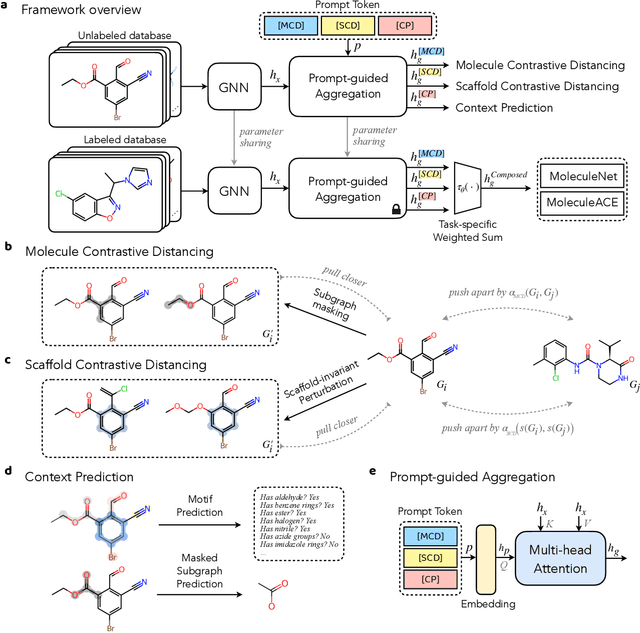
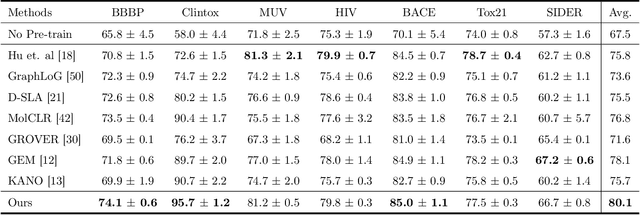
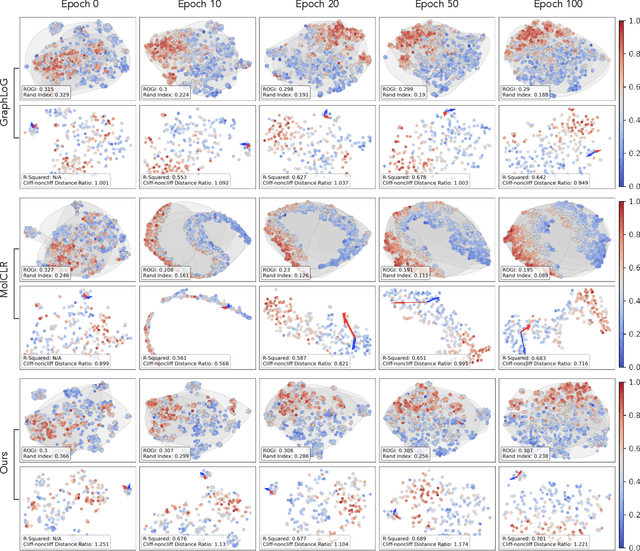

Reliable molecular property prediction is essential for various scientific endeavors and industrial applications, such as drug discovery. However, the scarcity of data, combined with the highly non-linear causal relationships between physicochemical and biological properties and conventional molecular featurization schemes, complicates the development of robust molecular machine learning models. Self-supervised learning (SSL) has emerged as a popular solution, utilizing large-scale, unannotated molecular data to learn a foundational representation of chemical space that might be advantageous for downstream tasks. Yet, existing molecular SSL methods largely overlook domain-specific knowledge, such as molecular similarity and scaffold importance, as well as the context of the target application when operating over the large chemical space. This paper introduces a novel learning framework that leverages the knowledge of structural hierarchies within molecular structures, embeds them through separate pre-training tasks over distinct channels, and employs a task-specific channel selection to compose a context-dependent representation. Our approach demonstrates competitive performance across various molecular property benchmarks and establishes some state-of-the-art results. It further offers unprecedented advantages in particularly challenging yet ubiquitous scenarios like activity cliffs with enhanced robustness and generalizability compared to other baselines.
Retroformer: Pushing the Limits of Interpretable End-to-end Retrosynthesis Transformer
Jan 29, 2022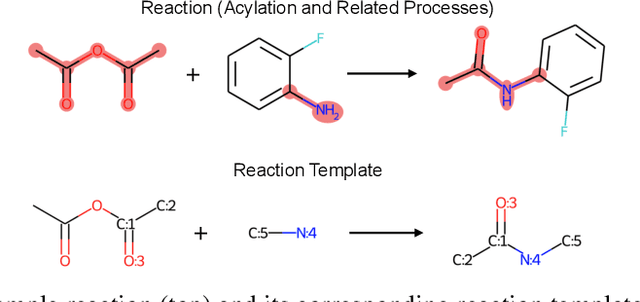
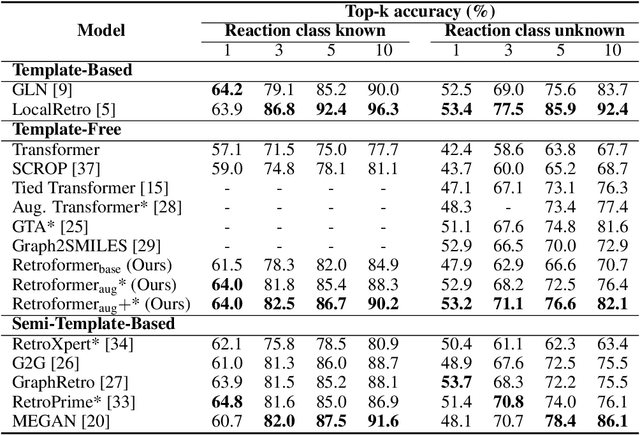
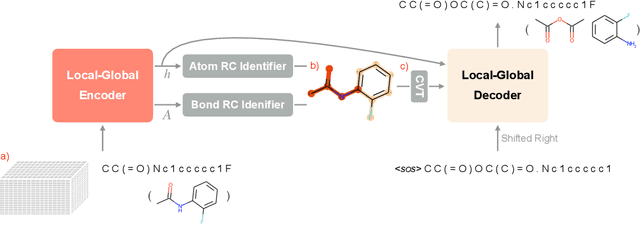
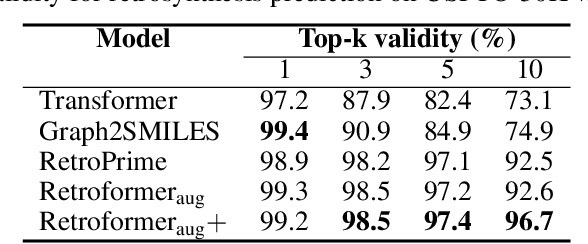
Retrosynthesis prediction is one of the fundamental challenges in organic synthesis. The task is to predict the reactants given a core product. With the advancement of machine learning, computer-aided synthesis planning has gained increasing interest. Numerous methods were proposed to solve this problem with different levels of dependency on additional chemical knowledge. In this paper, we propose Retroformer, a novel Transformer-based architecture for retrosynthesis prediction without relying on any cheminformatics tools for molecule editing. Via the proposed local attention head, the model can jointly encode the molecular sequence and graph, and efficiently exchange information between the local reactive region and the global reaction context. Retroformer reaches the new state-of-the-art accuracy for the end-to-end template-free retrosynthesis, and improves over many strong baselines on better molecule and reaction validity. In addition, its generative procedure is highly interpretable and controllable. Overall, Retroformer pushes the limits of the reaction reasoning ability of deep generative models.