 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRiNALMo: General-Purpose RNA Language Models Can Generalize Well on Structure Prediction Tasks
Feb 29, 2024Ribonucleic acid (RNA) plays a variety of crucial roles in fundamental biological processes. Recently, RNA has become an interesting drug target, emphasizing the need to improve our understanding of its structures and functions. Over the years, sequencing technologies have produced an enormous amount of unlabeled RNA data, which hides important knowledge and potential. Motivated by the successes of protein language models, we introduce RiboNucleic Acid Language Model (RiNALMo) to help unveil the hidden code of RNA. RiNALMo is the largest RNA language model to date with $650$ million parameters pre-trained on $36$ million non-coding RNA sequences from several available databases. RiNALMo is able to extract hidden knowledge and capture the underlying structure information implicitly embedded within the RNA sequences. RiNALMo achieves state-of-the-art results on several downstream tasks. Notably, we show that its generalization capabilities can overcome the inability of other deep learning methods for secondary structure prediction to generalize on unseen RNA families. The code has been made publicly available on https://github.com/lbcb-sci/RiNALMo.
Finding Hamiltonian cycles with graph neural networks
Jun 10, 2023We train a small message-passing graph neural network to predict Hamiltonian cycles on Erd\H{o}s-R\'enyi random graphs in a critical regime. It outperforms existing hand-crafted heuristics after about 2.5 hours of training on a single GPU. Our findings encourage an alternative approach to solving computationally demanding (NP-hard) problems arising in practice. Instead of devising a heuristic by hand, one can train it end-to-end using a neural network. This has several advantages. Firstly, it is relatively quick and requires little problem-specific knowledge. Secondly, the network can adjust to the distribution of training samples, improving the performance on the most relevant problem instances. The model is trained using supervised learning on artificially created problem instances; this training procedure does not use an existing solver to produce the supervised signal. Finally, the model generalizes well to larger graph sizes and retains reasonable performance even on graphs eight times the original size.
Learning to Untangle Genome Assembly with Graph Convolutional Networks
Jun 01, 2022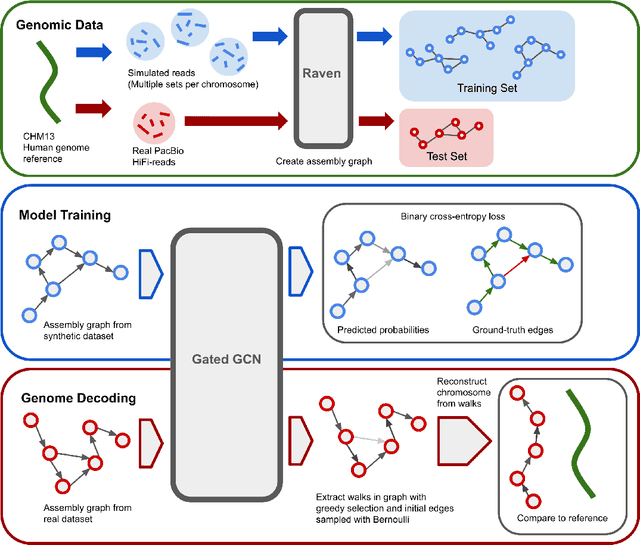
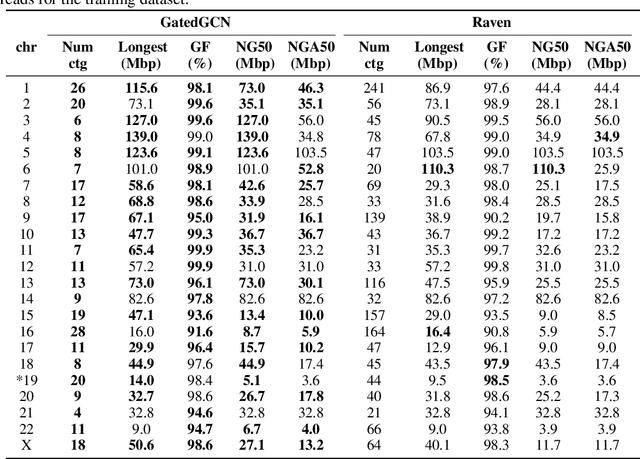
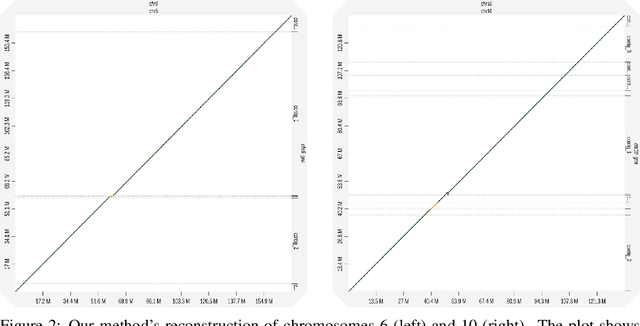
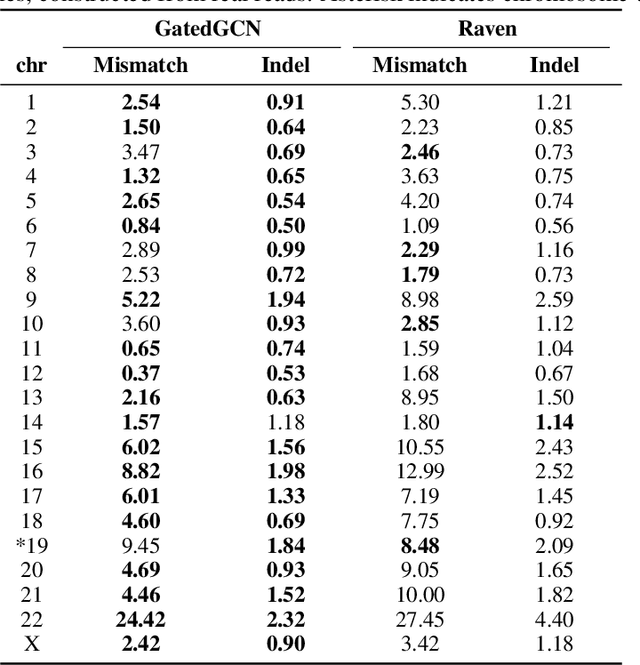
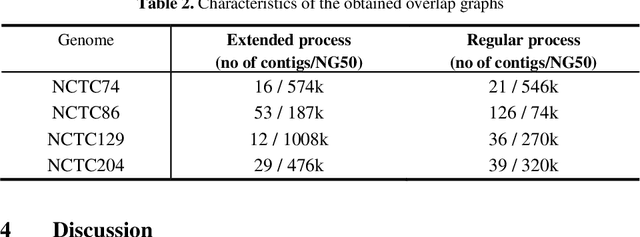
A quest to determine the complete sequence of a human DNA from telomere to telomere started three decades ago and was finally completed in 2021. This accomplishment was a result of a tremendous effort of numerous experts who engineered various tools and performed laborious manual inspection to achieve the first gapless genome sequence. However, such method can hardly be used as a general approach to assemble different genomes, especially when the assembly speed is critical given the large amount of data. In this work, we explore a different approach to the central part of the genome assembly task that consists of untangling a large assembly graph from which a genomic sequence needs to be reconstructed. Our main motivation is to reduce human-engineered heuristics and use deep learning to develop more generalizable reconstruction techniques. Precisely, we introduce a new learning framework to train a graph convolutional network to resolve assembly graphs by finding a correct path through them. The training is supervised with a dataset generated from the resolved CHM13 human sequence and tested on assembly graphs built using real human PacBio HiFi reads. Experimental results show that a model, trained on simulated graphs generated solely from a single chromosome, is able to remarkably resolve all other chromosomes. Moreover, the model outperforms hand-crafted heuristics from a state-of-the-art \textit{de novo} assembler on the same graphs. Reconstructed chromosomes with graph networks are more accurate on nucleotide level, report lower number of contigs, higher genome reconstructed fraction and NG50/NGA50 assessment metrics.
A step towards neural genome assembly
Nov 10, 2020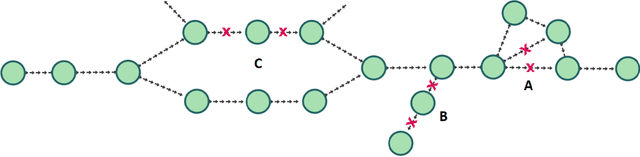
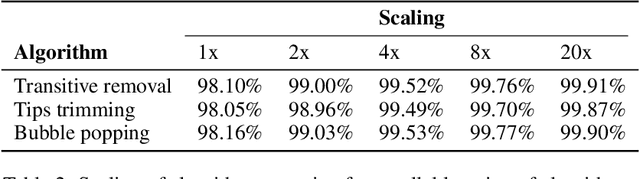
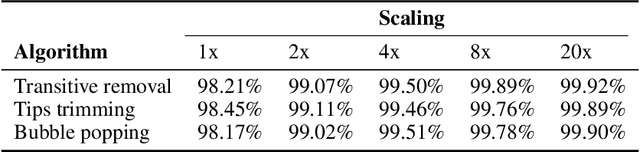

De novo genome assembly focuses on finding connections between a vast amount of short sequences in order to reconstruct the original genome. The central problem of genome assembly could be described as finding a Hamiltonian path through a large directed graph with a constraint that an unknown number of nodes and edges should be avoided. However, due to local structures in the graph and biological features, the problem can be reduced to graph simplification, which includes removal of redundant information. Motivated by recent advancements in graph representation learning and neural execution of algorithms, in this work we train the MPNN model with max-aggregator to execute several algorithms for graph simplification. We show that the algorithms were learned successfully and can be scaled to graphs of sizes up to 20 times larger than the ones used in training. We also test on graphs obtained from real-world genomic data---that of a lambda phage and E. coli.
Read classification using semi-supervised deep learning
Apr 23, 2019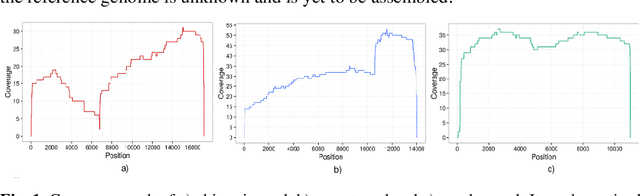
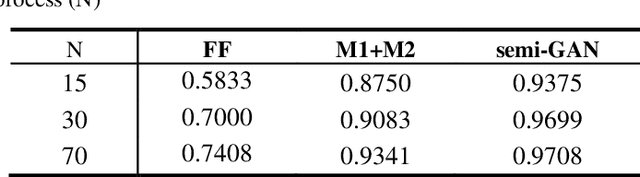
In this paper, we propose a semi-supervised deep learning method for detecting the specific types of reads that impede the de novo genome assembly process. Instead of dealing directly with sequenced reads, we analyze their coverage graphs converted to 1D-signals. We noticed that specific signal patterns occur in each relevant class of reads. Semi-supervised approach is chosen because manually labelling the data is a very slow and tedious process, so our goal was to facilitate the assembly process with as little labeled data as possible. We tested two models to learn patterns in the coverage graphs: M1+M2 and semi-GAN. We evaluated the performance of each model based on a manually labeled dataset that comprises various reads from multiple reference genomes with respect to the number of labeled examples that were used during the training process. In addition, we embedded our detection in the assembly process which improved the quality of assemblies.
MinCall - MinION end2end convolutional deep learning basecaller
Apr 22, 2019



The Oxford Nanopore Technologies's MinION is the first portable DNA sequencing device. It is capable of producing long reads, over 100 kBp were reported. However, it has significantly higher error rate than other methods. In this study, we present MinCall, an end2end basecaller model for the MinION. The model is based on deep learning and uses convolutional neural networks (CNN) in its implementation. For extra performance, it uses cutting edge deep learning techniques and architectures, batch normalization and Connectionist Temporal Classification (CTC) loss. The best performing deep learning model achieves 91.4% median match rate on E. Coli dataset using R9 pore chemistry and 1D reads.