 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAtomic Trajectory Modeling with State Space Models for Biomolecular Dynamics
Mar 18, 2026Understanding the dynamic behavior of biomolecules is fundamental to elucidating biological function and facilitating drug discovery. While Molecular Dynamics (MD) simulations provide a rigorous physical basis for studying these dynamics, they remain computationally expensive for long timescales. Conversely, recent deep generative models accelerate conformation generation but are typically either failing to model temporal relationship or built only for monomeric proteins. To bridge this gap, we introduce ATMOS, a novel generative framework based on State Space Models (SSM) designed to generate atom-level MD trajectories for biomolecular systems. ATMOS integrates a Pairformer-based state transition mechanism to capture long-range temporal dependencies, with a diffusion-based module to decode trajectory frames in an autoregressive manner. ATMOS is trained across crystal structures from PDB and conformation trajectory from large-scale MD simulation datasets including mdCATH and MISATO. We demonstrate that ATMOS achieves state-of-the-art performance in generating conformation trajectories for both protein monomers and complex protein-ligand systems. By enabling efficient inference of atomic trajectory of motions, this work establishes a promising foundation for modeling biomolecular dynamics.
ASTRA-bench: Evaluating Tool-Use Agent Reasoning and Action Planning with Personal User Context
Mar 02, 2026Next-generation AI must manage vast personal data, diverse tools, and multi-step reasoning, yet most benchmarks remain context-free and single-turn. We present ASTRA-bench (Assistant Skills in Tool-use, Reasoning \& Action-planning), a benchmark that uniquely unifies time-evolving personal context with an interactive toolbox and complex user intents. Our event-driven pipeline generates 2,413 scenarios across four protagonists, grounded in longitudinal life events and annotated by referential, functional, and informational complexity. Evaluation of state-of-the-art models (e.g., Claude-4.5-Opus, DeepSeek-V3.2) reveals significant performance degradation under high-complexity conditions, with argument generation emerging as the primary bottleneck. These findings expose critical limitations in current agents' ability to ground reasoning within messy personal context and orchestrate reliable multi-step plans. We release ASTRA-bench with a full execution environment and evaluation scripts to provide a diagnostic testbed for developing truly context-aware AI assistants.
Conditionally Site-Independent Neural Evolution of Antibody Sequences
Feb 21, 2026Common deep learning approaches for antibody engineering focus on modeling the marginal distribution of sequences. By treating sequences as independent samples, however, these methods overlook affinity maturation as a rich and largely untapped source of information about the evolutionary process by which antibodies explore the underlying fitness landscape. In contrast, classical phylogenetic models explicitly represent evolutionary dynamics but lack the expressivity to capture complex epistatic interactions. We bridge this gap with CoSiNE, a continuous-time Markov chain parameterized by a deep neural network. Mathematically, we prove that CoSiNE provides a first-order approximation to the intractable sequential point mutation process, capturing epistatic effects with an error bound that is quadratic in branch length. Empirically, CoSiNE outperforms state-of-the-art language models in zero-shot variant effect prediction by explicitly disentangling selection from context-dependent somatic hypermutation. Finally, we introduce Guided Gillespie, a classifier-guided sampling scheme that steers CoSiNE at inference time, enabling efficient optimization of antibody binding affinity toward specific antigens.
COMPASS: A Multi-Turn Benchmark for Tool-Mediated Planning & Preference Optimization
Oct 08, 2025
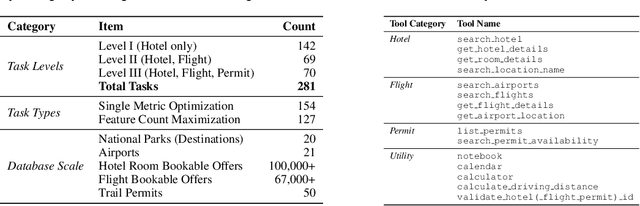
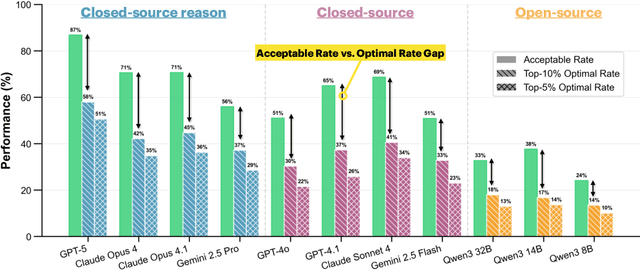

Real-world large language model (LLM) agents must master strategic tool use and user preference optimization through multi-turn interactions to assist users with complex planning tasks. We introduce COMPASS (Constrained Optimization through Multi-turn Planning and Strategic Solutions), a benchmark that evaluates agents on realistic travel-planning scenarios. We cast travel planning as a constrained preference optimization problem, where agents must satisfy hard constraints while simultaneously optimizing soft user preferences. To support this, we build a realistic travel database covering transportation, accommodation, and ticketing for 20 U.S. National Parks, along with a comprehensive tool ecosystem that mirrors commercial booking platforms. Evaluating state-of-the-art models, we uncover two critical gaps: (i) an acceptable-optimal gap, where agents reliably meet constraints but fail to optimize preferences, and (ii) a plan-coordination gap, where performance collapses on multi-service (flight and hotel) coordination tasks, especially for open-source models. By grounding reasoning and planning in a practical, user-facing domain, COMPASS provides a benchmark that directly measures an agent's ability to optimize user preferences in realistic tasks, bridging theoretical advances with real-world impact.
Measuring Scientific Capabilities of Language Models with a Systems Biology Dry Lab
Jul 02, 2025
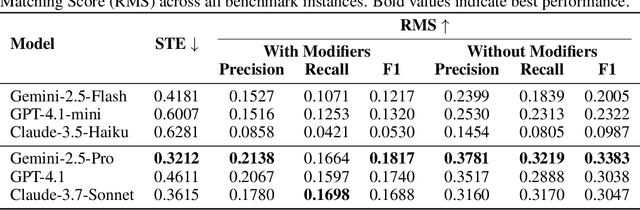
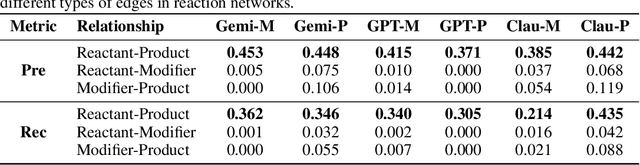
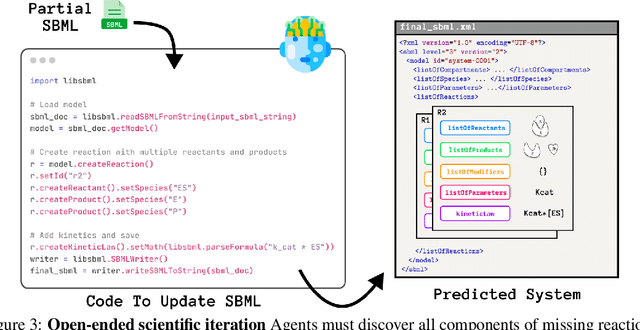
Designing experiments and result interpretations are core scientific competencies, particularly in biology, where researchers perturb complex systems to uncover the underlying systems. Recent efforts to evaluate the scientific capabilities of large language models (LLMs) fail to test these competencies because wet-lab experimentation is prohibitively expensive: in expertise, time and equipment. We introduce SciGym, a first-in-class benchmark that assesses LLMs' iterative experiment design and analysis abilities in open-ended scientific discovery tasks. SciGym overcomes the challenge of wet-lab costs by running a dry lab of biological systems. These models, encoded in Systems Biology Markup Language, are efficient for generating simulated data, making them ideal testbeds for experimentation on realistically complex systems. We evaluated six frontier LLMs on 137 small systems, and released a total of 350 systems. Our evaluation shows that while more capable models demonstrated superior performance, all models' performance declined significantly as system complexity increased, suggesting substantial room for improvement in the scientific capabilities of LLM agents.
Aligning Protein Conformation Ensemble Generation with Physical Feedback
May 30, 2025Protein dynamics play a crucial role in protein biological functions and properties, and their traditional study typically relies on time-consuming molecular dynamics (MD) simulations conducted in silico. Recent advances in generative modeling, particularly denoising diffusion models, have enabled efficient accurate protein structure prediction and conformation sampling by learning distributions over crystallographic structures. However, effectively integrating physical supervision into these data-driven approaches remains challenging, as standard energy-based objectives often lead to intractable optimization. In this paper, we introduce Energy-based Alignment (EBA), a method that aligns generative models with feedback from physical models, efficiently calibrating them to appropriately balance conformational states based on their energy differences. Experimental results on the MD ensemble benchmark demonstrate that EBA achieves state-of-the-art performance in generating high-quality protein ensembles. By improving the physical plausibility of generated structures, our approach enhances model predictions and holds promise for applications in structural biology and drug discovery.
Self-Evolving Curriculum for LLM Reasoning
May 20, 2025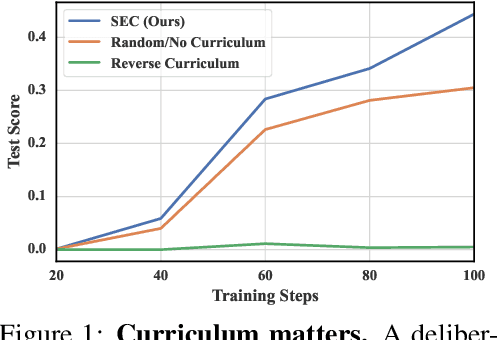
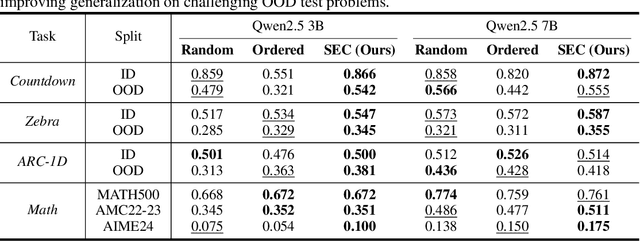
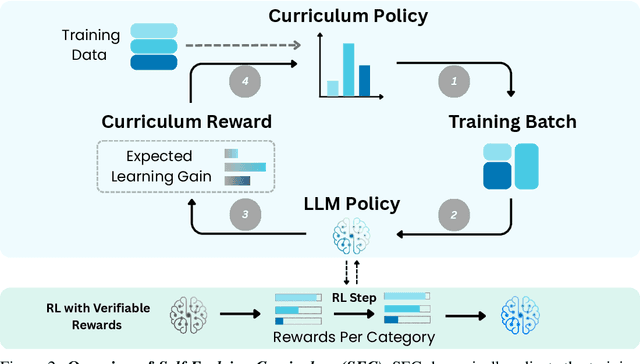
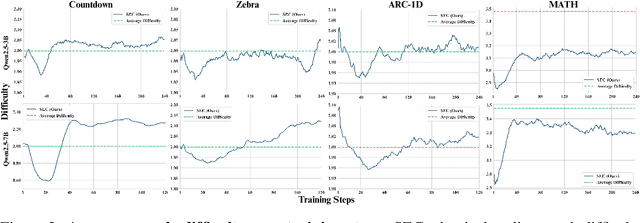
Reinforcement learning (RL) has proven effective for fine-tuning large language models (LLMs), significantly enhancing their reasoning abilities in domains such as mathematics and code generation. A crucial factor influencing RL fine-tuning success is the training curriculum: the order in which training problems are presented. While random curricula serve as common baselines, they remain suboptimal; manually designed curricula often rely heavily on heuristics, and online filtering methods can be computationally prohibitive. To address these limitations, we propose Self-Evolving Curriculum (SEC), an automatic curriculum learning method that learns a curriculum policy concurrently with the RL fine-tuning process. Our approach formulates curriculum selection as a non-stationary Multi-Armed Bandit problem, treating each problem category (e.g., difficulty level or problem type) as an individual arm. We leverage the absolute advantage from policy gradient methods as a proxy measure for immediate learning gain. At each training step, the curriculum policy selects categories to maximize this reward signal and is updated using the TD(0) method. Across three distinct reasoning domains: planning, inductive reasoning, and mathematics, our experiments demonstrate that SEC significantly improves models' reasoning capabilities, enabling better generalization to harder, out-of-distribution test problems. Additionally, our approach achieves better skill balance when fine-tuning simultaneously on multiple reasoning domains. These findings highlight SEC as a promising strategy for RL fine-tuning of LLMs.
Reaction-conditioned De Novo Enzyme Design with GENzyme
Nov 10, 2024The introduction of models like RFDiffusionAA, AlphaFold3, AlphaProteo, and Chai1 has revolutionized protein structure modeling and interaction prediction, primarily from a binding perspective, focusing on creating ideal lock-and-key models. However, these methods can fall short for enzyme-substrate interactions, where perfect binding models are rare, and induced fit states are more common. To address this, we shift to a functional perspective for enzyme design, where the enzyme function is defined by the reaction it catalyzes. Here, we introduce \textsc{GENzyme}, a \textit{de novo} enzyme design model that takes a catalytic reaction as input and generates the catalytic pocket, full enzyme structure, and enzyme-substrate binding complex. \textsc{GENzyme} is an end-to-end, three-staged model that integrates (1) a catalytic pocket generation and sequence co-design module, (2) a pocket inpainting and enzyme inverse folding module, and (3) a binding and screening module to optimize and predict enzyme-substrate complexes. The entire design process is driven by the catalytic reaction being targeted. This reaction-first approach allows for more accurate and biologically relevant enzyme design, potentially surpassing structure-based and binding-focused models in creating enzymes capable of catalyzing specific reactions. We provide \textsc{GENzyme} code at https://github.com/WillHua127/GENzyme.
Structure Language Models for Protein Conformation Generation
Oct 24, 2024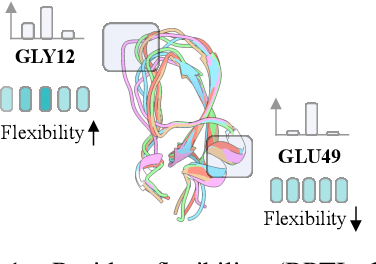
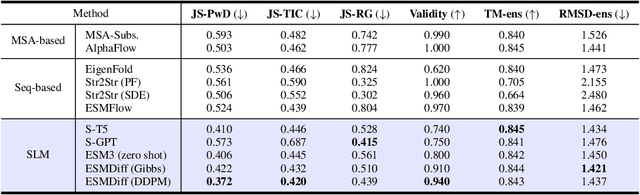
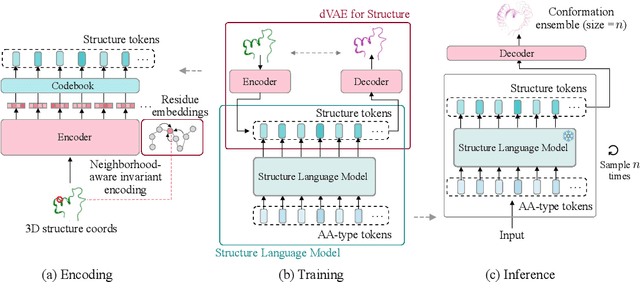
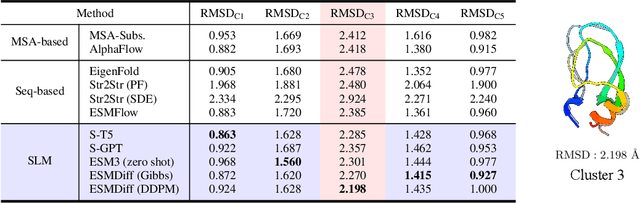
Proteins adopt multiple structural conformations to perform their diverse biological functions, and understanding these conformations is crucial for advancing drug discovery. Traditional physics-based simulation methods often struggle with sampling equilibrium conformations and are computationally expensive. Recently, deep generative models have shown promise in generating protein conformations as a more efficient alternative. However, these methods predominantly rely on the diffusion process within a 3D geometric space, which typically centers around the vicinity of metastable states and is often inefficient in terms of runtime. In this paper, we introduce Structure Language Modeling (SLM) as a novel framework for efficient protein conformation generation. Specifically, the protein structures are first encoded into a compact latent space using a discrete variational auto-encoder, followed by conditional language modeling that effectively captures sequence-specific conformation distributions. This enables a more efficient and interpretable exploration of diverse ensemble modes compared to existing methods. Based on this general framework, we instantiate SLM with various popular LM architectures as well as proposing the ESMDiff, a novel BERT-like structure language model fine-tuned from ESM3 with masked diffusion. We verify our approach in various scenarios, including the equilibrium dynamics of BPTI, conformational change pairs, and intrinsically disordered proteins. SLM provides a highly efficient solution, offering a 20-100x speedup than existing methods in generating diverse conformations, shedding light on promising avenues for future research.
ToolSandbox: A Stateful, Conversational, Interactive Evaluation Benchmark for LLM Tool Use Capabilities
Aug 08, 2024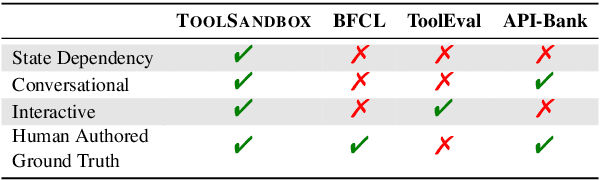
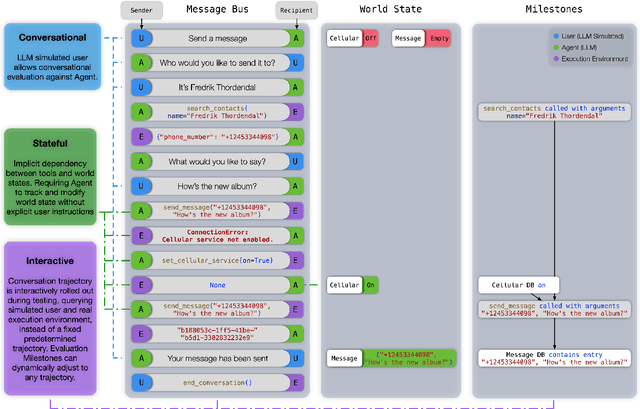
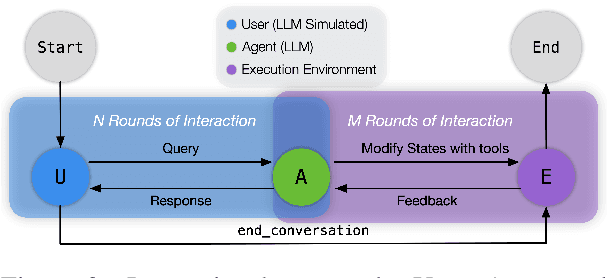
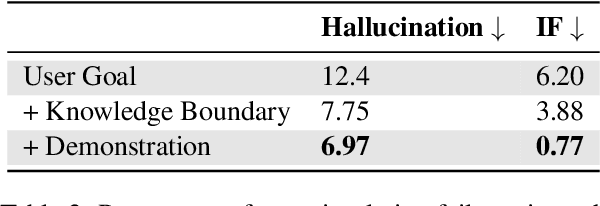
Recent large language models (LLMs) advancements sparked a growing research interest in tool assisted LLMs solving real-world challenges, which calls for comprehensive evaluation of tool-use capabilities. While previous works focused on either evaluating over stateless web services (RESTful API), based on a single turn user prompt, or an off-policy dialog trajectory, ToolSandbox includes stateful tool execution, implicit state dependencies between tools, a built-in user simulator supporting on-policy conversational evaluation and a dynamic evaluation strategy for intermediate and final milestones over an arbitrary trajectory. We show that open source and proprietary models have a significant performance gap, and complex tasks like State Dependency, Canonicalization and Insufficient Information defined in ToolSandbox are challenging even the most capable SOTA LLMs, providing brand-new insights into tool-use LLM capabilities. ToolSandbox evaluation framework is released at https://github.com/apple/ToolSandbox