 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRank-and-Reason: Multi-Agent Collaboration Accelerates Zero-Shot Protein Mutation Prediction
Feb 03, 2026Zero-shot mutation prediction is vital for low-resource protein engineering, yet existing protein language models (PLMs) often yield statistically confident results that ignore fundamental biophysical constraints. Currently, selecting candidates for wet-lab validation relies on manual expert auditing of PLM outputs, a process that is inefficient, subjective, and highly dependent on domain expertise. To address this, we propose Rank-and-Reason (VenusRAR), a two-stage agentic framework to automate this workflow and maximize expected wet-lab fitness. In the Rank-Stage, a Computational Expert and Virtual Biologist aggregate a context-aware multi-modal ensemble, establishing a new Spearman correlation record of 0.551 (vs. 0.518) on ProteinGym. In the Reason-Stage, an agentic Expert Panel employs chain-of-thought reasoning to audit candidates against geometric and structural constraints, improving the Top-5 Hit Rate by up to 367% on ProteinGym-DMS99. The wet-lab validation on Cas12i3 nuclease further confirms the framework's efficacy, achieving a 46.7% positive rate and identifying two novel mutants with 4.23-fold and 5.05-fold activity improvements. Code and datasets are released on GitHub (https://github.com/ai4protein/VenusRAR/).
VenusX: Unlocking Fine-Grained Functional Understanding of Proteins
May 17, 2025



Deep learning models have driven significant progress in predicting protein function and interactions at the protein level. While these advancements have been invaluable for many biological applications such as enzyme engineering and function annotation, a more detailed perspective is essential for understanding protein functional mechanisms and evaluating the biological knowledge captured by models. To address this demand, we introduce VenusX, the first large-scale benchmark for fine-grained functional annotation and function-based protein pairing at the residue, fragment, and domain levels. VenusX comprises three major task categories across six types of annotations, including residue-level binary classification, fragment-level multi-class classification, and pairwise functional similarity scoring for identifying critical active sites, binding sites, conserved sites, motifs, domains, and epitopes. The benchmark features over 878,000 samples curated from major open-source databases such as InterPro, BioLiP, and SAbDab. By providing mixed-family and cross-family splits at three sequence identity thresholds, our benchmark enables a comprehensive assessment of model performance on both in-distribution and out-of-distribution scenarios. For baseline evaluation, we assess a diverse set of popular and open-source models, including pre-trained protein language models, sequence-structure hybrids, structure-based methods, and alignment-based techniques. Their performance is reported across all benchmark datasets and evaluation settings using multiple metrics, offering a thorough comparison and a strong foundation for future research. Code and data are publicly available at https://github.com/ai4protein/VenusX.
Reactzyme: A Benchmark for Enzyme-Reaction Prediction
Aug 24, 2024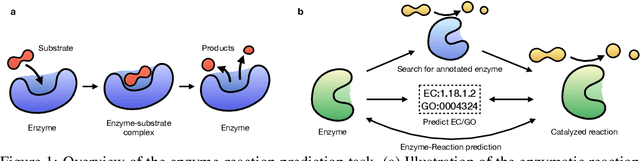
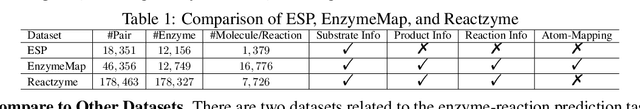
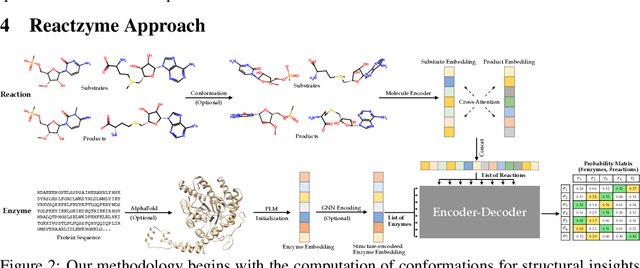
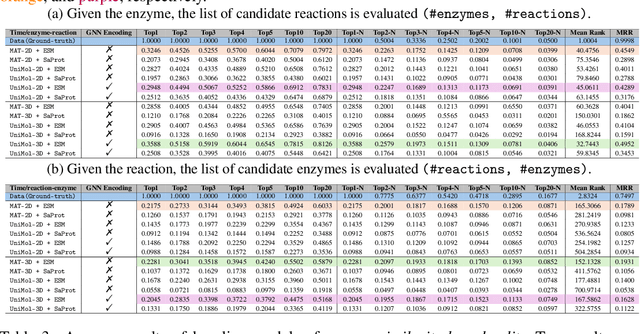
Enzymes, with their specific catalyzed reactions, are necessary for all aspects of life, enabling diverse biological processes and adaptations. Predicting enzyme functions is essential for understanding biological pathways, guiding drug development, enhancing bioproduct yields, and facilitating evolutionary studies. Addressing the inherent complexities, we introduce a new approach to annotating enzymes based on their catalyzed reactions. This method provides detailed insights into specific reactions and is adaptable to newly discovered reactions, diverging from traditional classifications by protein family or expert-derived reaction classes. We employ machine learning algorithms to analyze enzyme reaction datasets, delivering a much more refined view on the functionality of enzymes. Our evaluation leverages the largest enzyme-reaction dataset to date, derived from the SwissProt and Rhea databases with entries up to January 8, 2024. We frame the enzyme-reaction prediction as a retrieval problem, aiming to rank enzymes by their catalytic ability for specific reactions. With our model, we can recruit proteins for novel reactions and predict reactions in novel proteins, facilitating enzyme discovery and function annotation.
Autoregressive Enzyme Function Prediction with Multi-scale Multi-modality Fusion
Aug 11, 2024Accurate prediction of enzyme function is crucial for elucidating biological mechanisms and driving innovation across various sectors. Existing deep learning methods tend to rely solely on either sequence data or structural data and predict the EC number as a whole, neglecting the intrinsic hierarchical structure of EC numbers. To address these limitations, we introduce MAPred, a novel multi-modality and multi-scale model designed to autoregressively predict the EC number of proteins. MAPred integrates both the primary amino acid sequence and the 3D tokens of proteins, employing a dual-pathway approach to capture comprehensive protein characteristics and essential local functional sites. Additionally, MAPred utilizes an autoregressive prediction network to sequentially predict the digits of the EC number, leveraging the hierarchical organization of EC classifications. Evaluations on benchmark datasets, including New-392, Price, and New-815, demonstrate that our method outperforms existing models, marking a significant advance in the reliability and granularity of protein function prediction within bioinformatics.
Protein Representation Learning with Sequence Information Embedding: Does it Always Lead to a Better Performance?
Jun 28, 2024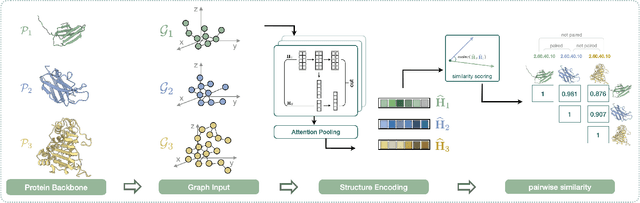
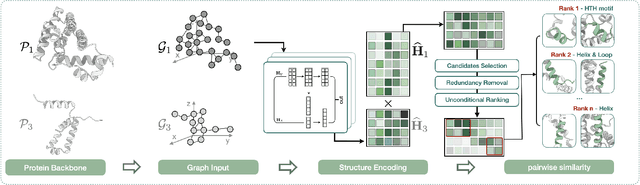

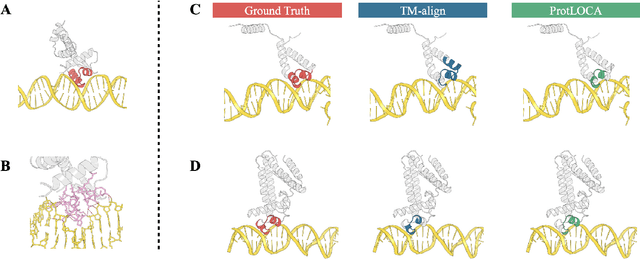
Deep learning has become a crucial tool in studying proteins. While the significance of modeling protein structure has been discussed extensively in the literature, amino acid types are typically included in the input as a default operation for many inference tasks. This study demonstrates with structure alignment task that embedding amino acid types in some cases may not help a deep learning model learn better representation. To this end, we propose ProtLOCA, a local geometry alignment method based solely on amino acid structure representation. The effectiveness of ProtLOCA is examined by a global structure-matching task on protein pairs with an independent test dataset based on CATH labels. Our method outperforms existing sequence- and structure-based representation learning methods by more quickly and accurately matching structurally consistent protein domains. Furthermore, in local structure pairing tasks, ProtLOCA for the first time provides a valid solution to highlight common local structures among proteins with different overall structures but the same function. This suggests a new possibility for using deep learning methods to analyze protein structure to infer function.
Simple, Efficient and Scalable Structure-aware Adapter Boosts Protein Language Models
Apr 23, 2024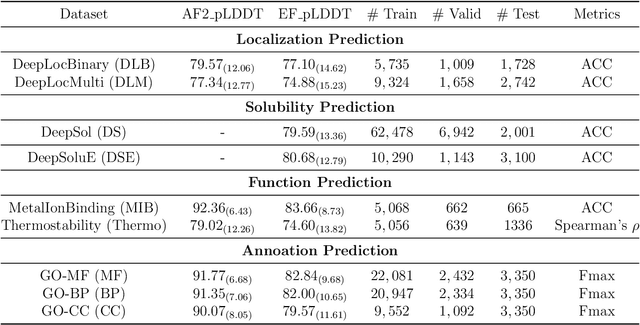
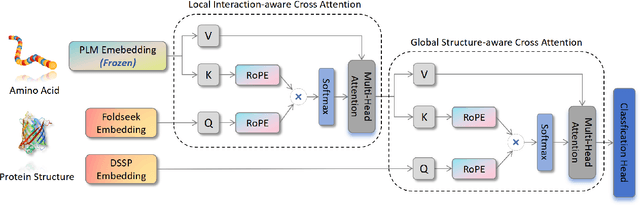
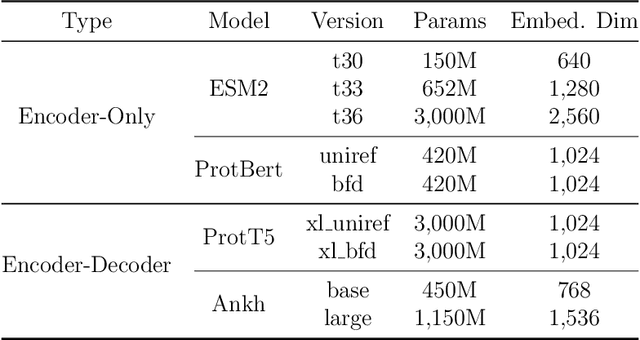
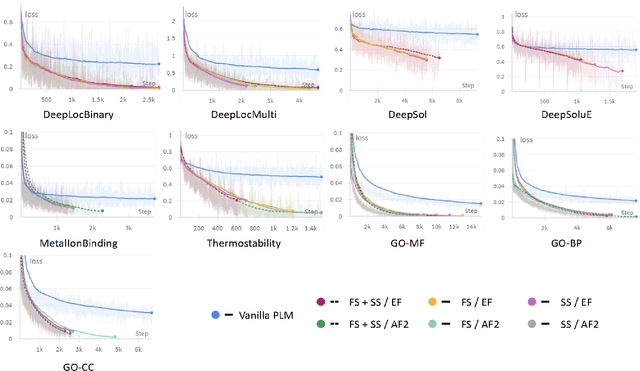
Fine-tuning Pre-trained protein language models (PLMs) has emerged as a prominent strategy for enhancing downstream prediction tasks, often outperforming traditional supervised learning approaches. As a widely applied powerful technique in natural language processing, employing Parameter-Efficient Fine-Tuning techniques could potentially enhance the performance of PLMs. However, the direct transfer to life science tasks is non-trivial due to the different training strategies and data forms. To address this gap, we introduce SES-Adapter, a simple, efficient, and scalable adapter method for enhancing the representation learning of PLMs. SES-Adapter incorporates PLM embeddings with structural sequence embeddings to create structure-aware representations. We show that the proposed method is compatible with different PLM architectures and across diverse tasks. Extensive evaluations are conducted on 2 types of folding structures with notable quality differences, 9 state-of-the-art baselines, and 9 benchmark datasets across distinct downstream tasks. Results show that compared to vanilla PLMs, SES-Adapter improves downstream task performance by a maximum of 11% and an average of 3%, with significantly accelerated training speed by a maximum of 1034% and an average of 362%, the convergence rate is also improved by approximately 2 times. Moreover, positive optimization is observed even with low-quality predicted structures. The source code for SES-Adapter is available at https://github.com/tyang816/SES-Adapter.
Fusing Neural and Physical: Augment Protein Conformation Sampling with Tractable Simulations
Feb 16, 2024The protein dynamics are common and important for their biological functions and properties, the study of which usually involves time-consuming molecular dynamics (MD) simulations in silico. Recently, generative models has been leveraged as a surrogate sampler to obtain conformation ensembles with orders of magnitude faster and without requiring any simulation data (a "zero-shot" inference). However, being agnostic of the underlying energy landscape, the accuracy of such generative model may still be limited. In this work, we explore the few-shot setting of such pre-trained generative sampler which incorporates MD simulations in a tractable manner. Specifically, given a target protein of interest, we first acquire some seeding conformations from the pre-trained sampler followed by a number of physical simulations in parallel starting from these seeding samples. Then we fine-tuned the generative model using the simulation trajectories above to become a target-specific sampler. Experimental results demonstrated the superior performance of such few-shot conformation sampler at a tractable computational cost.
PDB-Struct: A Comprehensive Benchmark for Structure-based Protein Design
Nov 30, 2023



Structure-based protein design has attracted increasing interest, with numerous methods being introduced in recent years. However, a universally accepted method for evaluation has not been established, since the wet-lab validation can be overly time-consuming for the development of new algorithms, and the $\textit{in silico}$ validation with recovery and perplexity metrics is efficient but may not precisely reflect true foldability. To address this gap, we introduce two novel metrics: refoldability-based metric, which leverages high-accuracy protein structure prediction models as a proxy for wet lab experiments, and stability-based metric, which assesses whether models can assign high likelihoods to experimentally stable proteins. We curate datasets from high-quality CATH protein data, high-throughput $\textit{de novo}$ designed proteins, and mega-scale experimental mutagenesis experiments, and in doing so, present the $\textbf{PDB-Struct}$ benchmark that evaluates both recent and previously uncompared protein design methods. Experimental results indicate that ByProt, ProteinMPNN, and ESM-IF perform exceptionally well on our benchmark, while ESM-Design and AF-Design fall short on the refoldability metric. We also show that while some methods exhibit high sequence recovery, they do not perform as well on our new benchmark. Our proposed benchmark paves the way for a fair and comprehensive evaluation of protein design methods in the future. Code is available at https://github.com/WANG-CR/PDB-Struct.
Score-based Enhanced Sampling for Protein Molecular Dynamics
Jun 05, 2023The dynamic nature of proteins is crucial for determining their biological functions and properties, and molecular dynamics (MD) simulations stand as a predominant tool to study such phenomena. By utilizing empirically derived force fields, MD simulations explore the conformational space through numerically evolving the system along MD trajectories. However, the high-energy barrier of the force fields can hamper the exploration of MD, resulting in inadequately sampled ensemble. In this paper, we propose leveraging score-based generative models (SGMs) trained on general protein structures to perform protein conformational sampling to complement traditional MD simulations. We argue that SGMs can provide a novel framework as an alternative to traditional enhanced sampling methods by learning multi-level score functions, which directly sample a diversity-controllable ensemble of conformations. We demonstrate the effectiveness of our approach on several benchmark systems by comparing the results with long MD trajectories and state-of-the-art generative structure prediction models. Our framework provides new insights that SGMs have the potential to serve as an efficient and simulation-free methods to study protein dynamics.
DiffPack: A Torsional Diffusion Model for Autoregressive Protein Side-Chain Packing
Jun 01, 2023Proteins play a critical role in carrying out biological functions, and their 3D structures are essential in determining their functions. Accurately predicting the conformation of protein side-chains given their backbones is important for applications in protein structure prediction, design and protein-protein interactions. Traditional methods are computationally intensive and have limited accuracy, while existing machine learning methods treat the problem as a regression task and overlook the restrictions imposed by the constant covalent bond lengths and angles. In this work, we present DiffPack, a torsional diffusion model that learns the joint distribution of side-chain torsional angles, the only degrees of freedom in side-chain packing, by diffusing and denoising on the torsional space. To avoid issues arising from simultaneous perturbation of all four torsional angles, we propose autoregressively generating the four torsional angles from \c{hi}1 to \c{hi}4 and training diffusion models for each torsional angle. We evaluate the method on several benchmarks for protein side-chain packing and show that our method achieves improvements of 11.9% and 13.5% in angle accuracy on CASP13 and CASP14, respectively, with a significantly smaller model size (60x fewer parameters). Additionally, we show the effectiveness of our method in enhancing side-chain predictions in the AlphaFold2 model. Code will be available upon the accept.