 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSelf-evolving AI agents for protein discovery and directed evolution
Mar 28, 2026Protein scientific discovery is bottlenecked by the manual orchestration of information and algorithms, while general agents are insufficient in complex domain projects. VenusFactory2 provides an autonomous framework that shifts from static tool usage to dynamic workflow synthesis via a self-evolving multi-agent infrastructure to address protein-related demands. It outperforms a set of well-known agents on the VenusAgentEval benchmark, and autonomously organizes the discovery and optimization of proteins from a single natural language prompt.
VenusX: Unlocking Fine-Grained Functional Understanding of Proteins
May 17, 2025



Deep learning models have driven significant progress in predicting protein function and interactions at the protein level. While these advancements have been invaluable for many biological applications such as enzyme engineering and function annotation, a more detailed perspective is essential for understanding protein functional mechanisms and evaluating the biological knowledge captured by models. To address this demand, we introduce VenusX, the first large-scale benchmark for fine-grained functional annotation and function-based protein pairing at the residue, fragment, and domain levels. VenusX comprises three major task categories across six types of annotations, including residue-level binary classification, fragment-level multi-class classification, and pairwise functional similarity scoring for identifying critical active sites, binding sites, conserved sites, motifs, domains, and epitopes. The benchmark features over 878,000 samples curated from major open-source databases such as InterPro, BioLiP, and SAbDab. By providing mixed-family and cross-family splits at three sequence identity thresholds, our benchmark enables a comprehensive assessment of model performance on both in-distribution and out-of-distribution scenarios. For baseline evaluation, we assess a diverse set of popular and open-source models, including pre-trained protein language models, sequence-structure hybrids, structure-based methods, and alignment-based techniques. Their performance is reported across all benchmark datasets and evaluation settings using multiple metrics, offering a thorough comparison and a strong foundation for future research. Code and data are publicly available at https://github.com/ai4protein/VenusX.
VenusFactory: A Unified Platform for Protein Engineering Data Retrieval and Language Model Fine-Tuning
Mar 19, 2025Natural language processing (NLP) has significantly influenced scientific domains beyond human language, including protein engineering, where pre-trained protein language models (PLMs) have demonstrated remarkable success. However, interdisciplinary adoption remains limited due to challenges in data collection, task benchmarking, and application. This work presents VenusFactory, a versatile engine that integrates biological data retrieval, standardized task benchmarking, and modular fine-tuning of PLMs. VenusFactory supports both computer science and biology communities with choices of both a command-line execution and a Gradio-based no-code interface, integrating $40+$ protein-related datasets and $40+$ popular PLMs. All implementations are open-sourced on https://github.com/tyang816/VenusFactory.
Retrieval-Enhanced Mutation Mastery: Augmenting Zero-Shot Prediction of Protein Language Model
Oct 28, 2024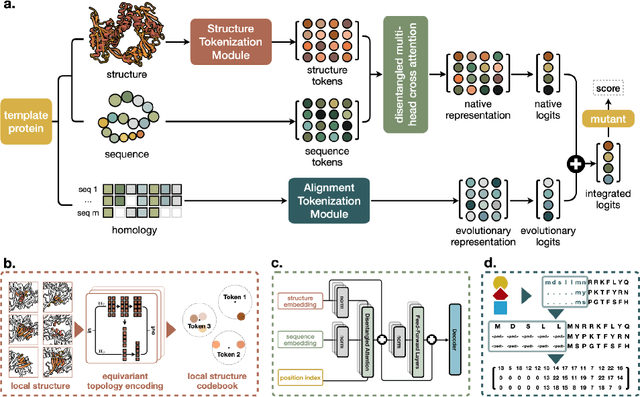
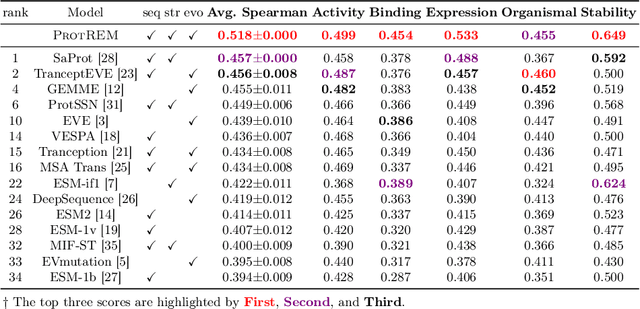
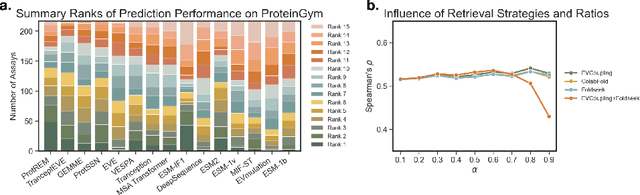
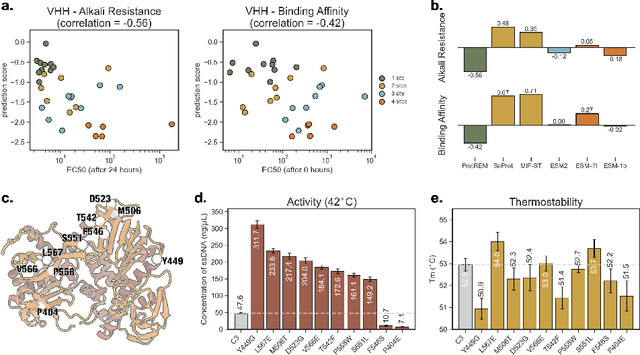
Enzyme engineering enables the modification of wild-type proteins to meet industrial and research demands by enhancing catalytic activity, stability, binding affinities, and other properties. The emergence of deep learning methods for protein modeling has demonstrated superior results at lower costs compared to traditional approaches such as directed evolution and rational design. In mutation effect prediction, the key to pre-training deep learning models lies in accurately interpreting the complex relationships among protein sequence, structure, and function. This study introduces a retrieval-enhanced protein language model for comprehensive analysis of native properties from sequence and local structural interactions, as well as evolutionary properties from retrieved homologous sequences. The state-of-the-art performance of the proposed ProtREM is validated on over 2 million mutants across 217 assays from an open benchmark (ProteinGym). We also conducted post-hoc analyses of the model's ability to improve the stability and binding affinity of a VHH antibody. Additionally, we designed 10 new mutants on a DNA polymerase and conducted wet-lab experiments to evaluate their enhanced activity at higher temperatures. Both in silico and experimental evaluations confirmed that our method provides reliable predictions of mutation effects, offering an auxiliary tool for biologists aiming to evolve existing enzymes. The implementation is publicly available at https://github.com/tyang816/ProtREM.
Immunogenicity Prediction with Dual Attention Enables Vaccine Target Selection
Oct 03, 2024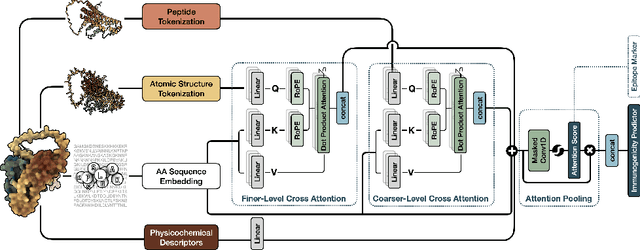
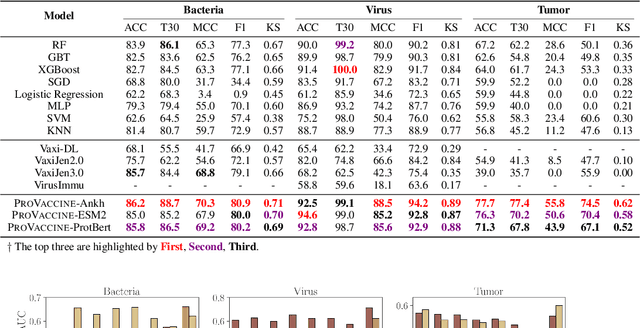
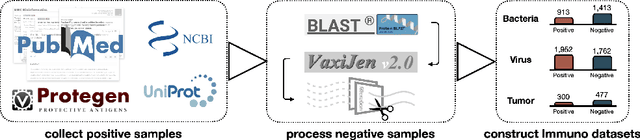

Immunogenicity prediction is a central topic in reverse vaccinology for finding candidate vaccines that can trigger protective immune responses. Existing approaches typically rely on highly compressed features and simple model architectures, leading to limited prediction accuracy and poor generalizability. To address these challenges, we introduce ProVaccine, a novel deep learning solution with a dual attention mechanism that integrates pre-trained latent vector representations of protein sequences and structures. We also compile the most comprehensive immunogenicity dataset to date, encompassing over 9,500 antigen sequences, structures, and immunogenicity labels from bacteria, viruses, and tumors. Extensive experiments demonstrate that ProVaccine outperforms existing methods across a wide range of evaluation metrics. Furthermore, we establish a post-hoc validation protocol to assess the practical significance of deep learning models in tackling vaccine design challenges. Our work provides an effective tool for vaccine design and sets valuable benchmarks for future research.
Secondary Structure-Guided Novel Protein Sequence Generation with Latent Graph Diffusion
Jul 10, 2024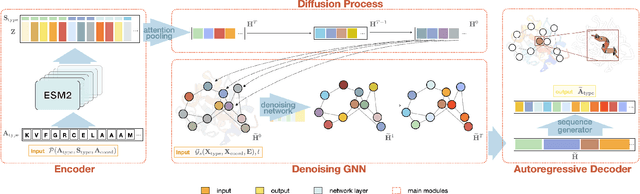
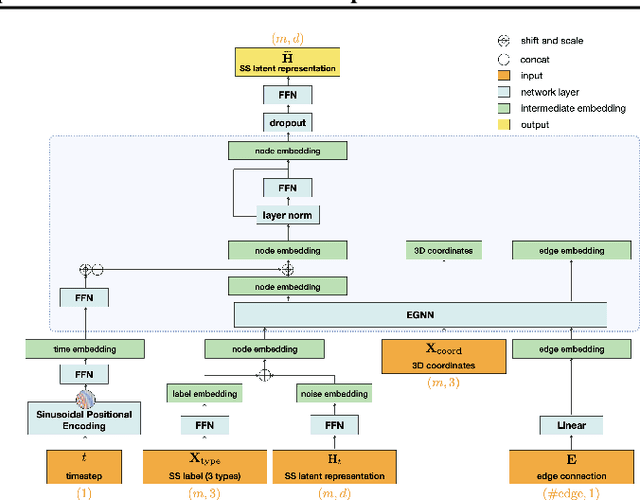

The advent of deep learning has introduced efficient approaches for de novo protein sequence design, significantly improving success rates and reducing development costs compared to computational or experimental methods. However, existing methods face challenges in generating proteins with diverse lengths and shapes while maintaining key structural features. To address these challenges, we introduce CPDiffusion-SS, a latent graph diffusion model that generates protein sequences based on coarse-grained secondary structural information. CPDiffusion-SS offers greater flexibility in producing a variety of novel amino acid sequences while preserving overall structural constraints, thus enhancing the reliability and diversity of generated proteins. Experimental analyses demonstrate the significant superiority of the proposed method in producing diverse and novel sequences, with CPDiffusion-SS surpassing popular baseline methods on open benchmarks across various quantitative measurements. Furthermore, we provide a series of case studies to highlight the biological significance of the generation performance by the proposed method. The source code is publicly available at https://github.com/riacd/CPDiffusion-SS
Protein Representation Learning with Sequence Information Embedding: Does it Always Lead to a Better Performance?
Jun 28, 2024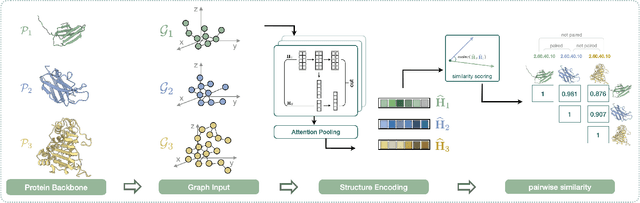
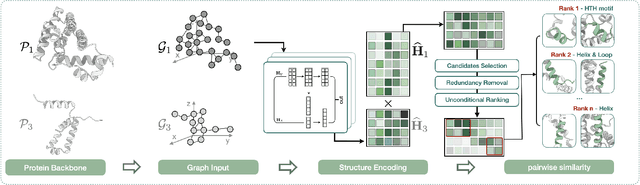

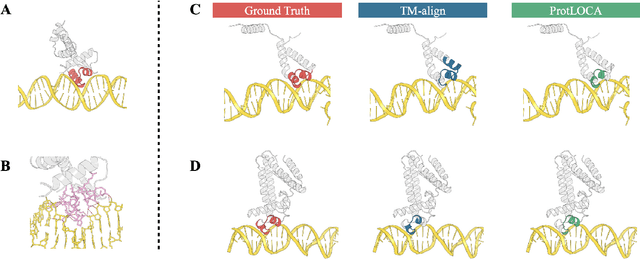
Deep learning has become a crucial tool in studying proteins. While the significance of modeling protein structure has been discussed extensively in the literature, amino acid types are typically included in the input as a default operation for many inference tasks. This study demonstrates with structure alignment task that embedding amino acid types in some cases may not help a deep learning model learn better representation. To this end, we propose ProtLOCA, a local geometry alignment method based solely on amino acid structure representation. The effectiveness of ProtLOCA is examined by a global structure-matching task on protein pairs with an independent test dataset based on CATH labels. Our method outperforms existing sequence- and structure-based representation learning methods by more quickly and accurately matching structurally consistent protein domains. Furthermore, in local structure pairing tasks, ProtLOCA for the first time provides a valid solution to highlight common local structures among proteins with different overall structures but the same function. This suggests a new possibility for using deep learning methods to analyze protein structure to infer function.
Simple, Efficient and Scalable Structure-aware Adapter Boosts Protein Language Models
Apr 23, 2024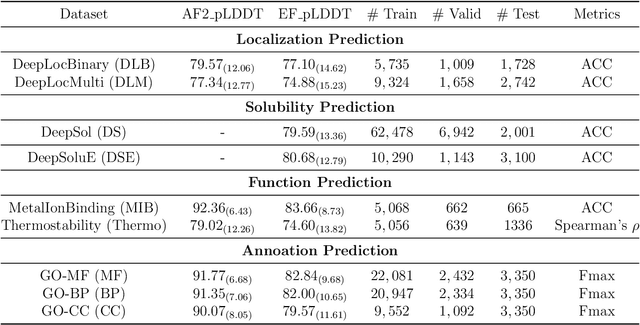
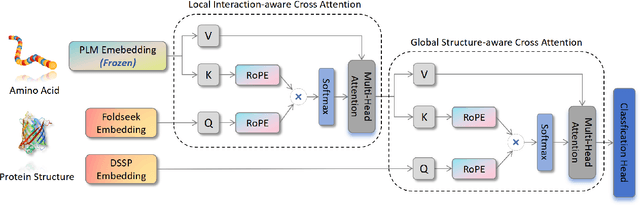
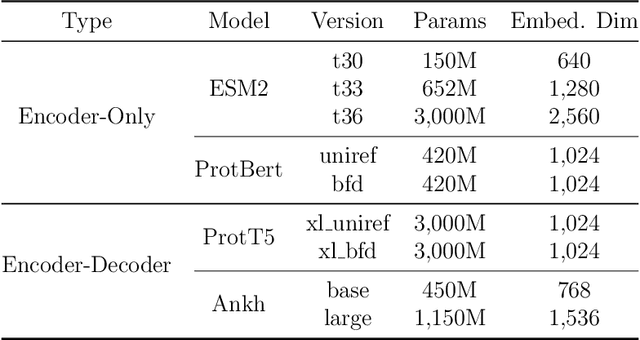
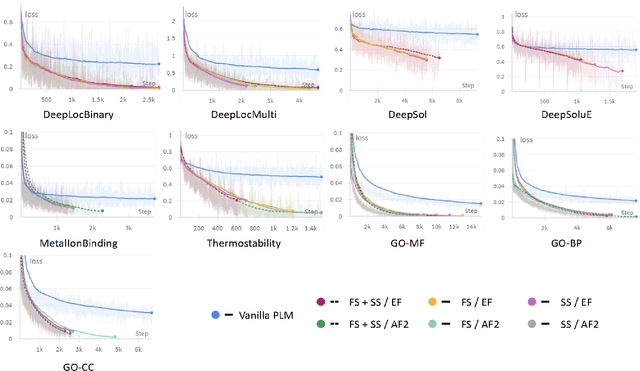
Fine-tuning Pre-trained protein language models (PLMs) has emerged as a prominent strategy for enhancing downstream prediction tasks, often outperforming traditional supervised learning approaches. As a widely applied powerful technique in natural language processing, employing Parameter-Efficient Fine-Tuning techniques could potentially enhance the performance of PLMs. However, the direct transfer to life science tasks is non-trivial due to the different training strategies and data forms. To address this gap, we introduce SES-Adapter, a simple, efficient, and scalable adapter method for enhancing the representation learning of PLMs. SES-Adapter incorporates PLM embeddings with structural sequence embeddings to create structure-aware representations. We show that the proposed method is compatible with different PLM architectures and across diverse tasks. Extensive evaluations are conducted on 2 types of folding structures with notable quality differences, 9 state-of-the-art baselines, and 9 benchmark datasets across distinct downstream tasks. Results show that compared to vanilla PLMs, SES-Adapter improves downstream task performance by a maximum of 11% and an average of 3%, with significantly accelerated training speed by a maximum of 1034% and an average of 362%, the convergence rate is also improved by approximately 2 times. Moreover, positive optimization is observed even with low-quality predicted structures. The source code for SES-Adapter is available at https://github.com/tyang816/SES-Adapter.
Two-stream joint matching method based on contrastive learning for few-shot action recognition
Jan 08, 2024Although few-shot action recognition based on metric learning paradigm has achieved significant success, it fails to address the following issues: (1) inadequate action relation modeling and underutilization of multi-modal information; (2) challenges in handling video matching problems with different lengths and speeds, and video matching problems with misalignment of video sub-actions. To address these issues, we propose a Two-Stream Joint Matching method based on contrastive learning (TSJM), which consists of two modules: Multi-modal Contrastive Learning Module (MCL) and Joint Matching Module (JMM). The objective of the MCL is to extensively investigate the inter-modal mutual information relationships, thereby thoroughly extracting modal information to enhance the modeling of action relationships. The JMM aims to simultaneously address the aforementioned video matching problems. The effectiveness of the proposed method is evaluated on two widely used few shot action recognition datasets, namely, SSv2 and Kinetics. Comprehensive ablation experiments are also conducted to substantiate the efficacy of our proposed approach.
A Unified View on Neural Message Passing with Opinion Dynamics for Social Networks
Oct 03, 2023Social networks represent a common form of interconnected data frequently depicted as graphs within the domain of deep learning-based inference. These communities inherently form dynamic systems, achieving stability through continuous internal communications and opinion exchanges among social actors along their social ties. In contrast, neural message passing in deep learning provides a clear and intuitive mathematical framework for understanding information propagation and aggregation among connected nodes in graphs. Node representations are dynamically updated by considering both the connectivity and status of neighboring nodes. This research harmonizes concepts from sociometry and neural message passing to analyze and infer the behavior of dynamic systems. Drawing inspiration from opinion dynamics in sociology, we propose ODNet, a novel message passing scheme incorporating bounded confidence, to refine the influence weight of local nodes for message propagation. We adjust the similarity cutoffs of bounded confidence and influence weights of ODNet and define opinion exchange rules that align with the characteristics of social network graphs. We show that ODNet enhances prediction performance across various graph types and alleviates oversmoothing issues. Furthermore, our approach surpasses conventional baselines in graph representation learning and proves its practical significance in analyzing real-world co-occurrence networks of metabolic genes. Remarkably, our method simplifies complex social network graphs solely by leveraging knowledge of interaction frequencies among entities within the system. It accurately identifies internal communities and the roles of genes in different metabolic pathways, including opinion leaders, bridge communicators, and isolators.