 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeImproved Offline Reinforcement Learning via Quantum Metric Encoding
Nov 13, 2025



Reinforcement learning (RL) with limited samples is common in real-world applications. However, offline RL performance under this constraint is often suboptimal. We consider an alternative approach to dealing with limited samples by introducing the Quantum Metric Encoder (QME). In this methodology, instead of applying the RL framework directly on the original states and rewards, we embed the states into a more compact and meaningful representation, where the structure of the encoding is inspired by quantum circuits. For classical data, QME is a classically simulable, trainable unitary embedding and thus serves as a quantum-inspired module, on a classical device. For quantum data in the form of quantum states, QME can be implemented directly on quantum hardware, allowing for training without measurement or re-encoding. We evaluated QME on three datasets, each limited to 100 samples. We use Soft-Actor-Critic (SAC) and Implicit-Q-Learning (IQL), two well-known RL algorithms, to demonstrate the effectiveness of our approach. From the experimental results, we find that training offline RL agents on QME-embedded states with decoded rewards yields significantly better performance than training on the original states and rewards. On average across the three datasets, for maximum reward performance, we achieve a 116.2% improvement for SAC and 117.6% for IQL. We further investigate the $Δ$-hyperbolicity of our framework, a geometric property of the state space known to be important for the RL training efficacy. The QME-embedded states exhibit low $Δ$-hyperbolicity, suggesting that the improvement after embedding arises from the modified geometry of the state space induced by QME. Thus, the low $Δ$-hyperbolicity and the corresponding effectiveness of QME could provide valuable information for developing efficient offline RL methods under limited-sample conditions.
A Unified View on Neural Message Passing with Opinion Dynamics for Social Networks
Oct 03, 2023Social networks represent a common form of interconnected data frequently depicted as graphs within the domain of deep learning-based inference. These communities inherently form dynamic systems, achieving stability through continuous internal communications and opinion exchanges among social actors along their social ties. In contrast, neural message passing in deep learning provides a clear and intuitive mathematical framework for understanding information propagation and aggregation among connected nodes in graphs. Node representations are dynamically updated by considering both the connectivity and status of neighboring nodes. This research harmonizes concepts from sociometry and neural message passing to analyze and infer the behavior of dynamic systems. Drawing inspiration from opinion dynamics in sociology, we propose ODNet, a novel message passing scheme incorporating bounded confidence, to refine the influence weight of local nodes for message propagation. We adjust the similarity cutoffs of bounded confidence and influence weights of ODNet and define opinion exchange rules that align with the characteristics of social network graphs. We show that ODNet enhances prediction performance across various graph types and alleviates oversmoothing issues. Furthermore, our approach surpasses conventional baselines in graph representation learning and proves its practical significance in analyzing real-world co-occurrence networks of metabolic genes. Remarkably, our method simplifies complex social network graphs solely by leveraging knowledge of interaction frequencies among entities within the system. It accurately identifies internal communities and the roles of genes in different metabolic pathways, including opinion leaders, bridge communicators, and isolators.
LLQL: Logistic Likelihood Q-Learning for Reinforcement Learning
Jul 05, 2023



Currently, research on Reinforcement learning (RL) can be broadly classified into two categories: online RL and offline RL. Both in online and offline RL, the primary focus of research on the Bellman error lies in the optimization techniques and performance improvement, rather than exploring the inherent structural properties of the Bellman error, such as distribution characteristics. In this study, we analyze the distribution of the Bellman approximation error in both online and offline settings. We find that in the online environment, the Bellman error follows a Logistic distribution, while in the offline environment, the Bellman error follows a constrained Logistic distribution, where the constrained distribution is dependent on the prior policy in the offline data set. Based on this finding, we have improved the MSELoss which is based on the assumption that the Bellman errors follow a normal distribution, and we utilized the Logistic maximum likelihood function to construct $\rm LLoss$ as an alternative loss function. In addition, we observed that the rewards in the offline data set should follow a specific distribution, which would facilitate the achievement of offline objectives. In our numerical experiments, we performed controlled variable corrections on the loss functions of two variants of Soft-Actor-Critic in both online and offline environments. The results confirmed our hypothesis regarding the online and offline settings, we also found that the variance of LLoss is smaller than MSELoss. Our research provides valuable insights for further investigations based on the distribution of Bellman errors.
Accurate and Definite Mutational Effect Prediction with Lightweight Equivariant Graph Neural Networks
Apr 13, 2023
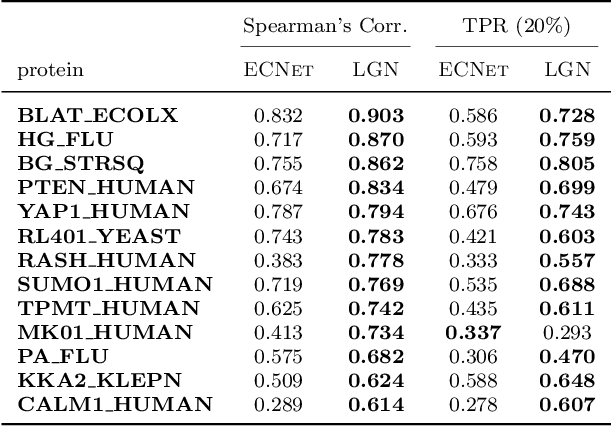
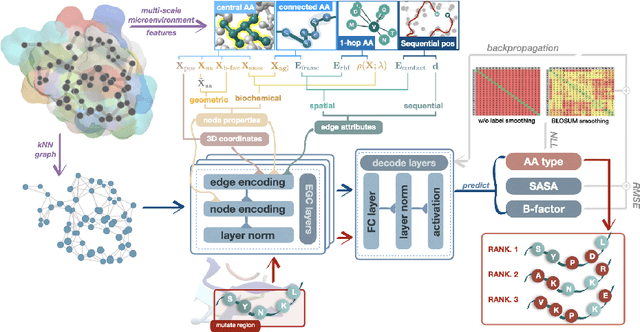

Directed evolution as a widely-used engineering strategy faces obstacles in finding desired mutants from the massive size of candidate modifications. While deep learning methods learn protein contexts to establish feasible searching space, many existing models are computationally demanding and fail to predict how specific mutational tests will affect a protein's sequence or function. This research introduces a lightweight graph representation learning scheme that efficiently analyzes the microenvironment of wild-type proteins and recommends practical higher-order mutations exclusive to the user-specified protein and function of interest. Our method enables continuous improvement of the inference model by limited computational resources and a few hundred mutational training samples, resulting in accurate prediction of variant effects that exhibit near-perfect correlation with the ground truth across deep mutational scanning assays of 19 proteins. With its affordability and applicability to both computer scientists and biochemical laboratories, our solution offers a wide range of benefits that make it an ideal choice for the community.