 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeAccurate and Definite Mutational Effect Prediction with Lightweight Equivariant Graph Neural Networks
Paper and Code

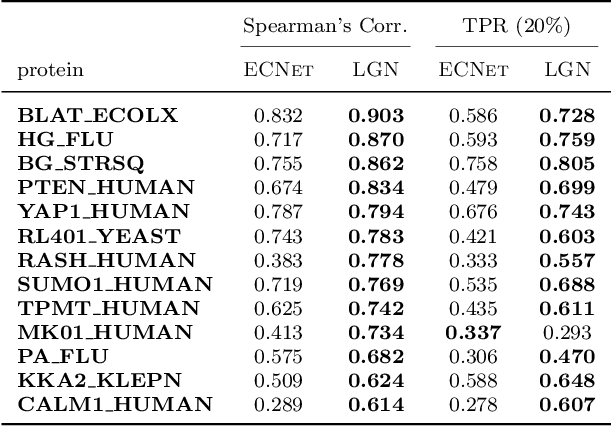
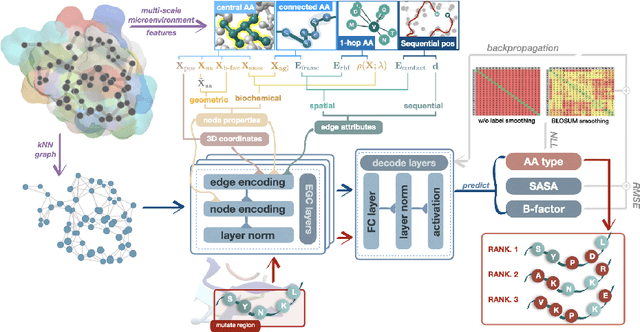

Directed evolution as a widely-used engineering strategy faces obstacles in finding desired mutants from the massive size of candidate modifications. While deep learning methods learn protein contexts to establish feasible searching space, many existing models are computationally demanding and fail to predict how specific mutational tests will affect a protein's sequence or function. This research introduces a lightweight graph representation learning scheme that efficiently analyzes the microenvironment of wild-type proteins and recommends practical higher-order mutations exclusive to the user-specified protein and function of interest. Our method enables continuous improvement of the inference model by limited computational resources and a few hundred mutational training samples, resulting in accurate prediction of variant effects that exhibit near-perfect correlation with the ground truth across deep mutational scanning assays of 19 proteins. With its affordability and applicability to both computer scientists and biochemical laboratories, our solution offers a wide range of benefits that make it an ideal choice for the community.