 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSCP: Accelerating Discovery with a Global Web of Autonomous Scientific Agents
Dec 30, 2025We introduce SCP: the Science Context Protocol, an open-source standard designed to accelerate discovery by enabling a global network of autonomous scientific agents. SCP is built on two foundational pillars: (1) Unified Resource Integration: At its core, SCP provides a universal specification for describing and invoking scientific resources, spanning software tools, models, datasets, and physical instruments. This protocol-level standardization enables AI agents and applications to discover, call, and compose capabilities seamlessly across disparate platforms and institutional boundaries. (2) Orchestrated Experiment Lifecycle Management: SCP complements the protocol with a secure service architecture, which comprises a centralized SCP Hub and federated SCP Servers. This architecture manages the complete experiment lifecycle (registration, planning, execution, monitoring, and archival), enforces fine-grained authentication and authorization, and orchestrates traceable, end-to-end workflows that bridge computational and physical laboratories. Based on SCP, we have constructed a scientific discovery platform that offers researchers and agents a large-scale ecosystem of more than 1,600 tool resources. Across diverse use cases, SCP facilitates secure, large-scale collaboration between heterogeneous AI systems and human researchers while significantly reducing integration overhead and enhancing reproducibility. By standardizing scientific context and tool orchestration at the protocol level, SCP establishes essential infrastructure for scalable, multi-institution, agent-driven science.
COMET: Benchmark for Comprehensive Biological Multi-omics Evaluation Tasks and Language Models
Dec 13, 2024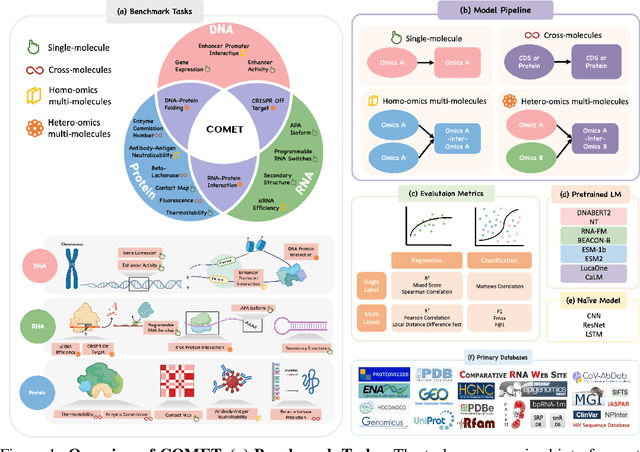
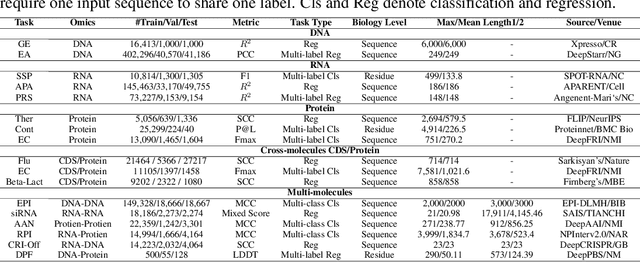
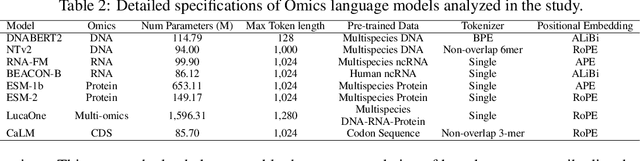
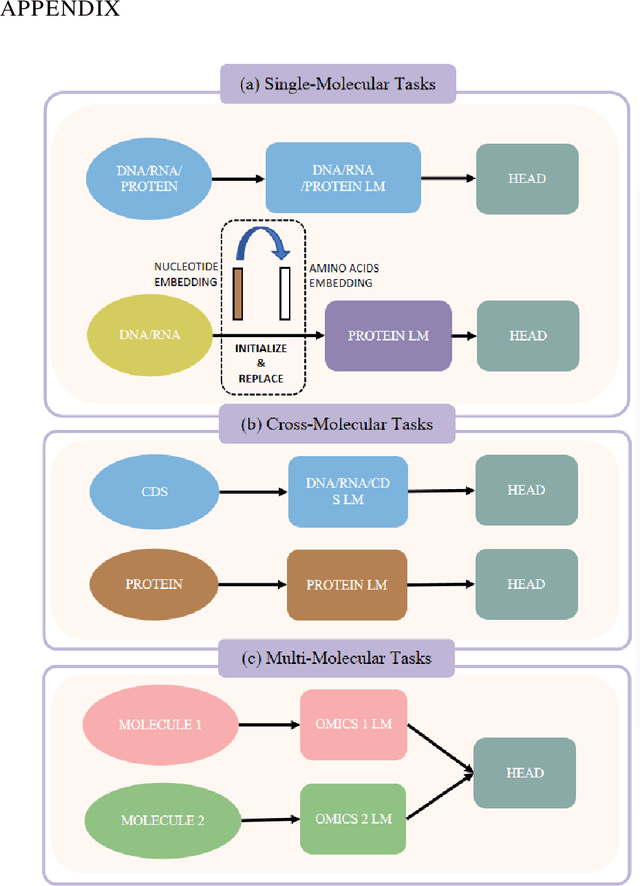
As key elements within the central dogma, DNA, RNA, and proteins play crucial roles in maintaining life by guaranteeing accurate genetic expression and implementation. Although research on these molecules has profoundly impacted fields like medicine, agriculture, and industry, the diversity of machine learning approaches-from traditional statistical methods to deep learning models and large language models-poses challenges for researchers in choosing the most suitable models for specific tasks, especially for cross-omics and multi-omics tasks due to the lack of comprehensive benchmarks. To address this, we introduce the first comprehensive multi-omics benchmark COMET (Benchmark for Biological COmprehensive Multi-omics Evaluation Tasks and Language Models), designed to evaluate models across single-omics, cross-omics, and multi-omics tasks. First, we curate and develop a diverse collection of downstream tasks and datasets covering key structural and functional aspects in DNA, RNA, and proteins, including tasks that span multiple omics levels. Then, we evaluate existing foundational language models for DNA, RNA, and proteins, as well as the newly proposed multi-omics method, offering valuable insights into their performance in integrating and analyzing data from different biological modalities. This benchmark aims to define critical issues in multi-omics research and guide future directions, ultimately promoting advancements in understanding biological processes through integrated and different omics data analysis.
Simple, Efficient and Scalable Structure-aware Adapter Boosts Protein Language Models
Apr 23, 2024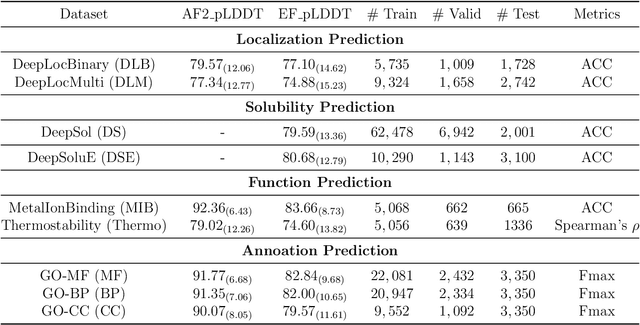
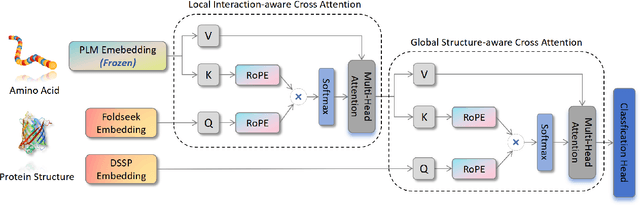
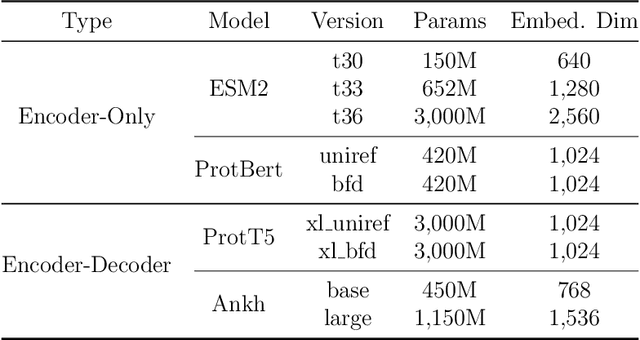
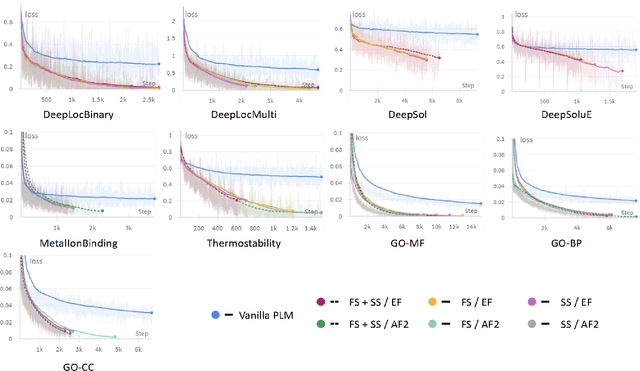
Fine-tuning Pre-trained protein language models (PLMs) has emerged as a prominent strategy for enhancing downstream prediction tasks, often outperforming traditional supervised learning approaches. As a widely applied powerful technique in natural language processing, employing Parameter-Efficient Fine-Tuning techniques could potentially enhance the performance of PLMs. However, the direct transfer to life science tasks is non-trivial due to the different training strategies and data forms. To address this gap, we introduce SES-Adapter, a simple, efficient, and scalable adapter method for enhancing the representation learning of PLMs. SES-Adapter incorporates PLM embeddings with structural sequence embeddings to create structure-aware representations. We show that the proposed method is compatible with different PLM architectures and across diverse tasks. Extensive evaluations are conducted on 2 types of folding structures with notable quality differences, 9 state-of-the-art baselines, and 9 benchmark datasets across distinct downstream tasks. Results show that compared to vanilla PLMs, SES-Adapter improves downstream task performance by a maximum of 11% and an average of 3%, with significantly accelerated training speed by a maximum of 1034% and an average of 362%, the convergence rate is also improved by approximately 2 times. Moreover, positive optimization is observed even with low-quality predicted structures. The source code for SES-Adapter is available at https://github.com/tyang816/SES-Adapter.
PETA: Evaluating the Impact of Protein Transfer Learning with Sub-word Tokenization on Downstream Applications
Oct 26, 2023

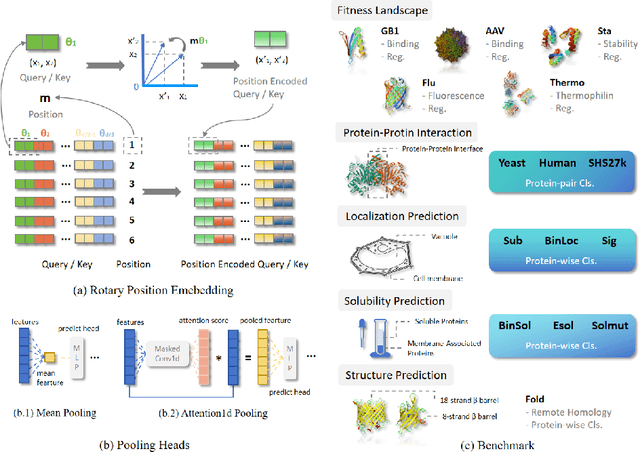
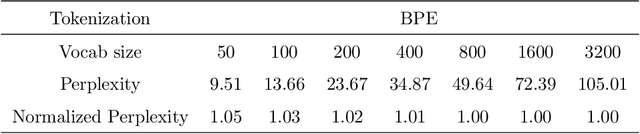
Large protein language models are adept at capturing the underlying evolutionary information in primary structures, offering significant practical value for protein engineering. Compared to natural language models, protein amino acid sequences have a smaller data volume and a limited combinatorial space. Choosing an appropriate vocabulary size to optimize the pre-trained model is a pivotal issue. Moreover, despite the wealth of benchmarks and studies in the natural language community, there remains a lack of a comprehensive benchmark for systematically evaluating protein language model quality. Given these challenges, PETA trained language models with 14 different vocabulary sizes under three tokenization methods. It conducted thousands of tests on 33 diverse downstream datasets to assess the models' transfer learning capabilities, incorporating two classification heads and three random seeds to mitigate potential biases. Extensive experiments indicate that vocabulary sizes between 50 and 200 optimize the model, whereas sizes exceeding 800 detrimentally affect the model's representational performance. Our code, model weights and datasets are available at https://github.com/ginnm/ProteinPretraining.
Accurate and Definite Mutational Effect Prediction with Lightweight Equivariant Graph Neural Networks
Apr 13, 2023
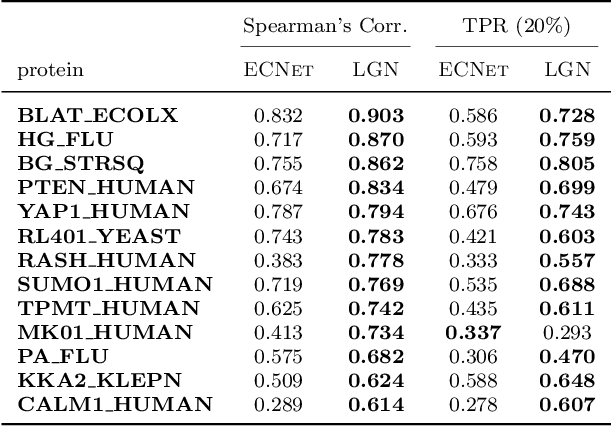
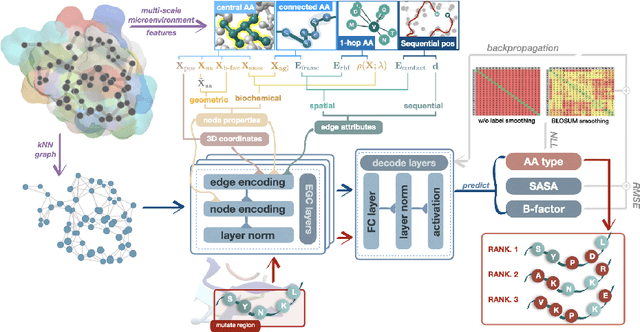

Directed evolution as a widely-used engineering strategy faces obstacles in finding desired mutants from the massive size of candidate modifications. While deep learning methods learn protein contexts to establish feasible searching space, many existing models are computationally demanding and fail to predict how specific mutational tests will affect a protein's sequence or function. This research introduces a lightweight graph representation learning scheme that efficiently analyzes the microenvironment of wild-type proteins and recommends practical higher-order mutations exclusive to the user-specified protein and function of interest. Our method enables continuous improvement of the inference model by limited computational resources and a few hundred mutational training samples, resulting in accurate prediction of variant effects that exhibit near-perfect correlation with the ground truth across deep mutational scanning assays of 19 proteins. With its affordability and applicability to both computer scientists and biochemical laboratories, our solution offers a wide range of benefits that make it an ideal choice for the community.
SESNet: sequence-structure feature-integrated deep learning method for data-efficient protein engineering
Dec 29, 2022Deep learning has been widely used for protein engineering. However, it is limited by the lack of sufficient experimental data to train an accurate model for predicting the functional fitness of high-order mutants. Here, we develop SESNet, a supervised deep-learning model to predict the fitness for protein mutants by leveraging both sequence and structure information, and exploiting attention mechanism. Our model integrates local evolutionary context from homologous sequences, the global evolutionary context encoding rich semantic from the universal protein sequence space and the structure information accounting for the microenvironment around each residue in a protein. We show that SESNet outperforms state-of-the-art models for predicting the sequence-function relationship on 26 deep mutational scanning datasets. More importantly, we propose a data augmentation strategy by leveraging the data from unsupervised models to pre-train our model. After that, our model can achieve strikingly high accuracy in prediction of the fitness of protein mutants, especially for the higher order variants (> 4 mutation sites), when finetuned by using only a small number of experimental mutation data (<50). The strategy proposed is of great practical value as the required experimental effort, i.e., producing a few tens of experimental mutation data on a given protein, is generally affordable by an ordinary biochemical group and can be applied on almost any protein.
InterFace:Adjustable Angular Margin Inter-class Loss for Deep Face Recognition
Oct 09, 2022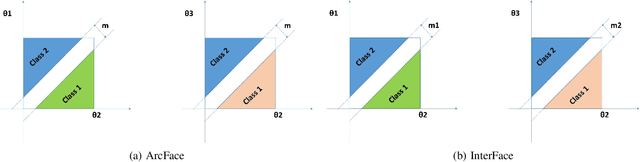
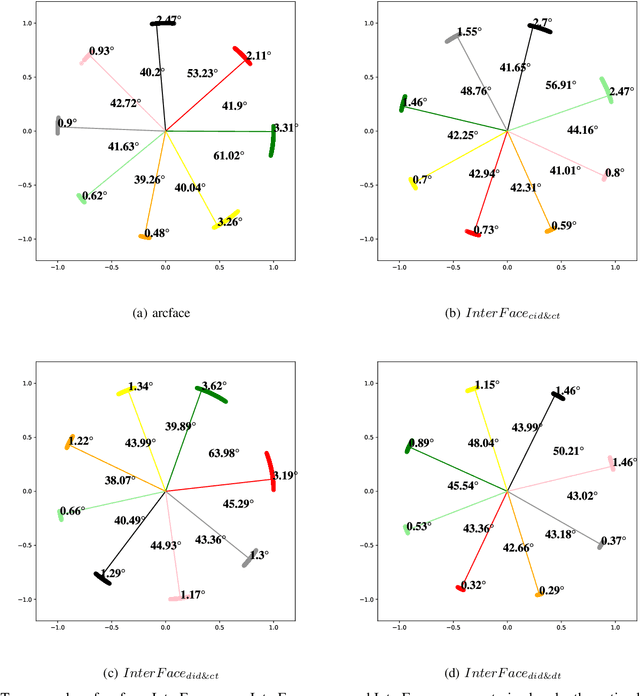
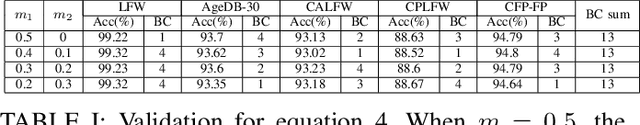
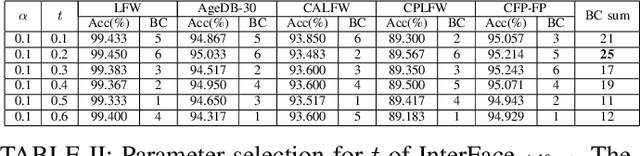
In the field of face recognition, it is always a hot research topic to improve the loss solution to make the face features extracted by the network have greater discriminative power. Research works in recent years has improved the discriminative power of the face model by normalizing softmax to the cosine space step by step and then adding a fixed penalty margin to reduce the intra-class distance to increase the inter-class distance. Although a great deal of previous work has been done to optimize the boundary penalty to improve the discriminative power of the model, adding a fixed margin penalty to the depth feature and the corresponding weight is not consistent with the pattern of data in the real scenario. To address this issue, in this paper, we propose a novel loss function, InterFace, releasing the constraint of adding a margin penalty only between the depth feature and the corresponding weight to push the separability of classes by adding corresponding margin penalties between the depth features and all weights. To illustrate the advantages of InterFace over a fixed penalty margin, we explained geometrically and comparisons on a set of mainstream benchmarks. From a wider perspective, our InterFace has advanced the state-of-the-art face recognition performance on five out of thirteen mainstream benchmarks. All training codes, pre-trained models, and training logs, are publicly released \footnote{$https://github.com/iamsangmeng/InterFace$}.