 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeInference-Time Alignment Control for Diffusion Models with Reinforcement Learning Guidance
Aug 28, 2025Denoising-based generative models, particularly diffusion and flow matching algorithms, have achieved remarkable success. However, aligning their output distributions with complex downstream objectives, such as human preferences, compositional accuracy, or data compressibility, remains challenging. While reinforcement learning (RL) fine-tuning methods, inspired by advances in RL from human feedback (RLHF) for large language models, have been adapted to these generative frameworks, current RL approaches are suboptimal for diffusion models and offer limited flexibility in controlling alignment strength after fine-tuning. In this work, we reinterpret RL fine-tuning for diffusion models through the lens of stochastic differential equations and implicit reward conditioning. We introduce Reinforcement Learning Guidance (RLG), an inference-time method that adapts Classifier-Free Guidance (CFG) by combining the outputs of the base and RL fine-tuned models via a geometric average. Our theoretical analysis shows that RLG's guidance scale is mathematically equivalent to adjusting the KL-regularization coefficient in standard RL objectives, enabling dynamic control over the alignment-quality trade-off without further training. Extensive experiments demonstrate that RLG consistently improves the performance of RL fine-tuned models across various architectures, RL algorithms, and downstream tasks, including human preferences, compositional control, compressibility, and text rendering. Furthermore, RLG supports both interpolation and extrapolation, thereby offering unprecedented flexibility in controlling generative alignment. Our approach provides a practical and theoretically sound solution for enhancing and controlling diffusion model alignment at inference. The source code for RLG is publicly available at the Github: https://github.com/jinluo12345/Reinforcement-learning-guidance.
Model Decides How to Tokenize: Adaptive DNA Sequence Tokenization with MxDNA
Dec 18, 2024Foundation models have made significant strides in understanding the genomic language of DNA sequences. However, previous models typically adopt the tokenization methods designed for natural language, which are unsuitable for DNA sequences due to their unique characteristics. In addition, the optimal approach to tokenize DNA remains largely under-explored, and may not be intuitively understood by humans even if discovered. To address these challenges, we introduce MxDNA, a novel framework where the model autonomously learns an effective DNA tokenization strategy through gradient decent. MxDNA employs a sparse Mixture of Convolution Experts coupled with a deformable convolution to model the tokenization process, with the discontinuous, overlapping, and ambiguous nature of meaningful genomic segments explicitly considered. On Nucleotide Transformer Benchmarks and Genomic Benchmarks, MxDNA demonstrates superior performance to existing methods with less pretraining data and time, highlighting its effectiveness. Finally, we show that MxDNA learns unique tokenization strategy distinct to those of previous methods and captures genomic functionalities at a token level during self-supervised pretraining. Our MxDNA aims to provide a new perspective on DNA tokenization, potentially offering broad applications in various domains and yielding profound insights.
COMET: Benchmark for Comprehensive Biological Multi-omics Evaluation Tasks and Language Models
Dec 13, 2024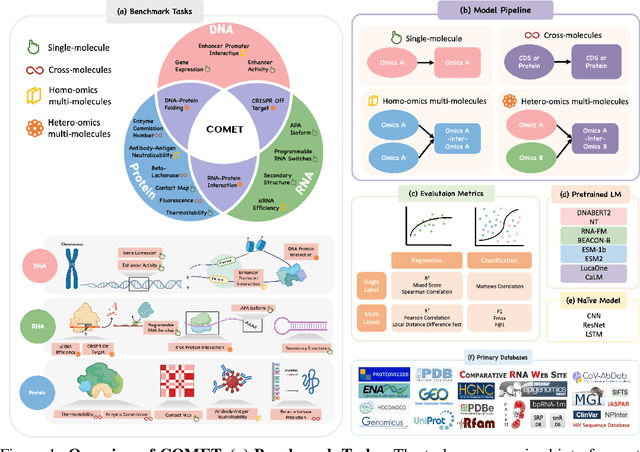
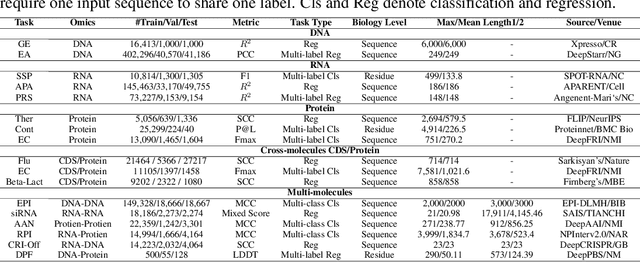
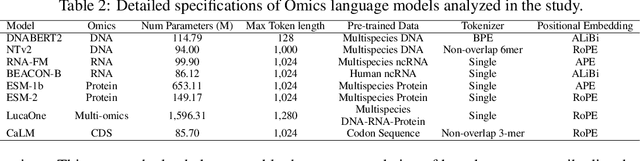
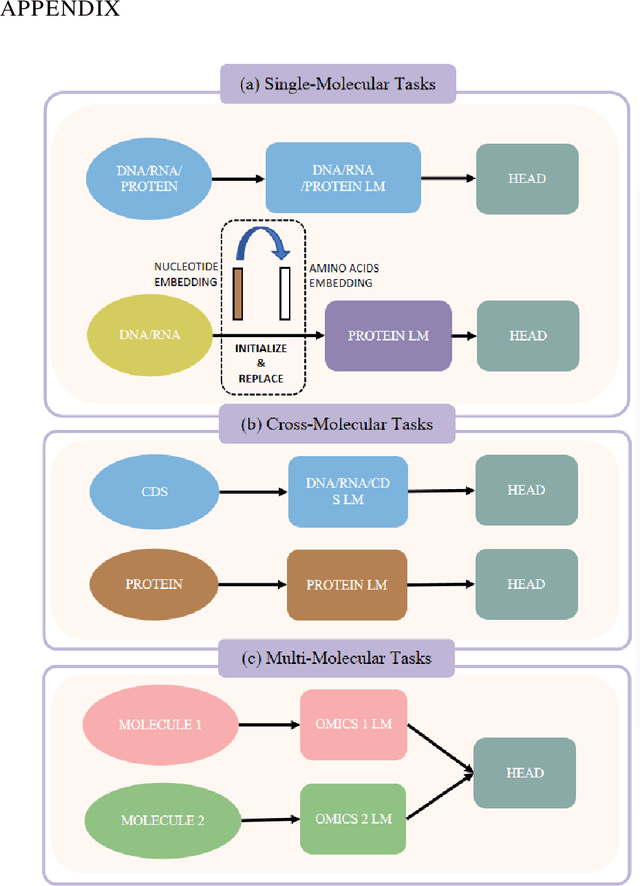
As key elements within the central dogma, DNA, RNA, and proteins play crucial roles in maintaining life by guaranteeing accurate genetic expression and implementation. Although research on these molecules has profoundly impacted fields like medicine, agriculture, and industry, the diversity of machine learning approaches-from traditional statistical methods to deep learning models and large language models-poses challenges for researchers in choosing the most suitable models for specific tasks, especially for cross-omics and multi-omics tasks due to the lack of comprehensive benchmarks. To address this, we introduce the first comprehensive multi-omics benchmark COMET (Benchmark for Biological COmprehensive Multi-omics Evaluation Tasks and Language Models), designed to evaluate models across single-omics, cross-omics, and multi-omics tasks. First, we curate and develop a diverse collection of downstream tasks and datasets covering key structural and functional aspects in DNA, RNA, and proteins, including tasks that span multiple omics levels. Then, we evaluate existing foundational language models for DNA, RNA, and proteins, as well as the newly proposed multi-omics method, offering valuable insights into their performance in integrating and analyzing data from different biological modalities. This benchmark aims to define critical issues in multi-omics research and guide future directions, ultimately promoting advancements in understanding biological processes through integrated and different omics data analysis.
LayerMatch: Do Pseudo-labels Benefit All Layers?
Jun 20, 2024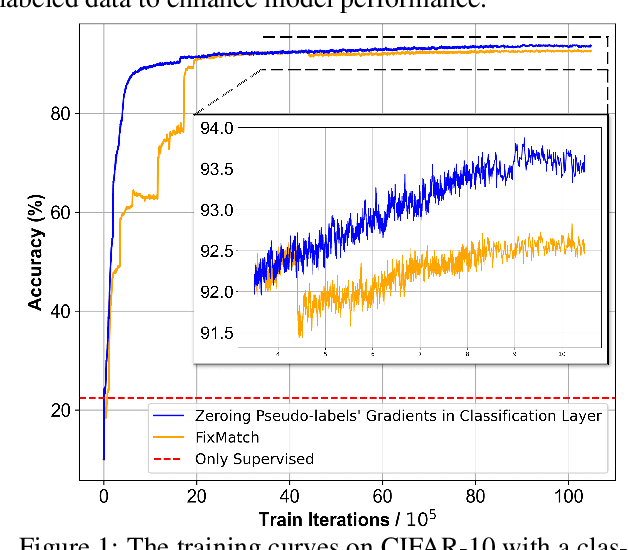
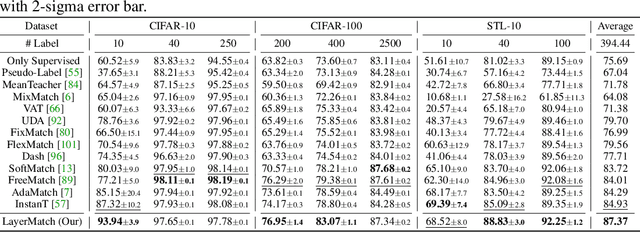
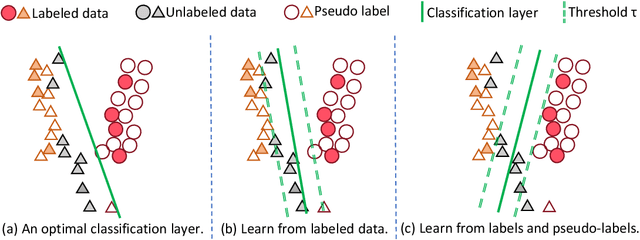
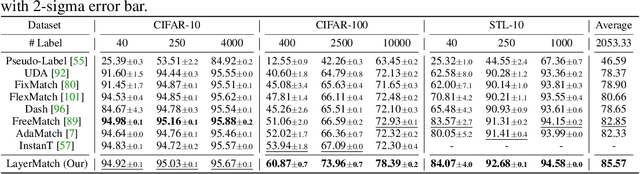
Deep neural networks have achieved remarkable performance across various tasks when supplied with large-scale labeled data. However, the collection of labeled data can be time-consuming and labor-intensive. Semi-supervised learning (SSL), particularly through pseudo-labeling algorithms that iteratively assign pseudo-labels for self-training, offers a promising solution to mitigate the dependency of labeled data. Previous research generally applies a uniform pseudo-labeling strategy across all model layers, assuming that pseudo-labels exert uniform influence throughout. Contrasting this, our theoretical analysis and empirical experiment demonstrate feature extraction layer and linear classification layer have distinct learning behaviors in response to pseudo-labels. Based on these insights, we develop two layer-specific pseudo-label strategies, termed Grad-ReLU and Avg-Clustering. Grad-ReLU mitigates the impact of noisy pseudo-labels by removing the gradient detrimental effects of pseudo-labels in the linear classification layer. Avg-Clustering accelerates the convergence of feature extraction layer towards stable clustering centers by integrating consistent outputs. Our approach, LayerMatch, which integrates these two strategies, can avoid the severe interference of noisy pseudo-labels in the linear classification layer while accelerating the clustering capability of the feature extraction layer. Through extensive experimentation, our approach consistently demonstrates exceptional performance on standard semi-supervised learning benchmarks, achieving a significant improvement of 10.38% over baseline method and a 2.44% increase compared to state-of-the-art methods.
BEACON: Benchmark for Comprehensive RNA Tasks and Language Models
Jun 14, 2024RNA plays a pivotal role in translating genetic instructions into functional outcomes, underscoring its importance in biological processes and disease mechanisms. Despite the emergence of numerous deep learning approaches for RNA, particularly universal RNA language models, there remains a significant lack of standardized benchmarks to assess the effectiveness of these methods. In this study, we introduce the first comprehensive RNA benchmark BEACON (\textbf{BE}nchm\textbf{A}rk for \textbf{CO}mprehensive R\textbf{N}A Task and Language Models). First, BEACON comprises 13 distinct tasks derived from extensive previous work covering structural analysis, functional studies, and engineering applications, enabling a comprehensive assessment of the performance of methods on various RNA understanding tasks. Second, we examine a range of models, including traditional approaches like CNNs, as well as advanced RNA foundation models based on language models, offering valuable insights into the task-specific performances of these models. Third, we investigate the vital RNA language model components from the tokenizer and positional encoding aspects. Notably, our findings emphasize the superiority of single nucleotide tokenization and the effectiveness of Attention with Linear Biases (ALiBi) over traditional positional encoding methods. Based on these insights, a simple yet strong baseline called BEACON-B is proposed, which can achieve outstanding performance with limited data and computational resources. The datasets and source code of our benchmark are available at https://github.com/terry-r123/RNABenchmark.
Rethinking the BERT-like Pretraining for DNA Sequences
Oct 12, 2023With the success of large-scale pretraining in NLP, there is an increasing trend of applying it to the domain of life sciences. In particular, pretraining methods based on DNA sequences have garnered growing attention due to their potential to capture generic information about genes. However, existing pretraining methods for DNA sequences largely rely on direct adoptions of BERT pretraining from NLP, lacking a comprehensive understanding and a specifically tailored approach. To address this research gap, we first conducted a series of exploratory experiments and gained several insightful observations: 1) In the fine-tuning phase of downstream tasks, when using K-mer overlapping tokenization instead of K-mer non-overlapping tokenization, both overlapping and non-overlapping pretraining weights show consistent performance improvement.2) During the pre-training process, using K-mer overlapping tokenization quickly produces clear K-mer embeddings and reduces the loss to a very low level, while using K-mer non-overlapping tokenization results in less distinct embeddings and continuously decreases the loss. 3) Using overlapping tokenization causes the self-attention in the intermediate layers of pre-trained models to tend to overly focus on certain tokens, reflecting that these layers are not adequately optimized. In summary, overlapping tokenization can benefit the fine-tuning of downstream tasks but leads to inadequate pretraining with fast convergence. To unleash the pretraining potential, we introduce a novel approach called RandomMask, which gradually increases the task difficulty of BERT-like pretraining by continuously expanding its mask boundary, forcing the model to learn more knowledge. RandomMask is simple but effective, achieving top-tier performance across 26 datasets of 28 datasets spanning 7 downstream tasks.