 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRiskFlow: Fast and Faithful Safety-Critical Traffic Scenario Generation
Jun 04, 2026Safety-critical traffic scenario generation is essential for evaluating autonomous driving systems under rare but high-risk interactions. Existing diffusion-based methods offer strong controllability in closed-loop generation, but their iterative denoising process is computationally expensive and may accumulate sampling and guidance errors over long rollouts, causing unrealistic motion artifacts such as jitter, abnormal acceleration, and off-road behavior. To address these issues, we propose RiskFlow, a closed-loop safety-critical multi-agent traffic generation framework that formulates future trajectory generation as transport in the action space. Instead of relying on iterative denoising, RiskFlow learns an average velocity field over a finite interval to transform Gaussian action sequences into future acceleration and yaw-rate commands with a single forward pass, using a JVP-based objective for efficient and stable training. At test time, RiskFlow applies output-space guidance to the generated actions, steering selected critical agents toward risky interactions while regularizing off-road behavior, and reconstructs physically feasible trajectories through vehicle dynamics. Experiments on nuScenes with tbsim closed-loop evaluation show that RiskFlow achieves a strong adversariality-realism trade-off across multi-agent and long-horizon settings. Compared with representative baselines, RiskFlow consistently improves realism while maintaining competitive safety-critical generation capability, and substantially reduces inference time for evaluation.
Biology Instructions: A Dataset and Benchmark for Multi-Omics Sequence Understanding Capability of Large Language Models
Dec 26, 2024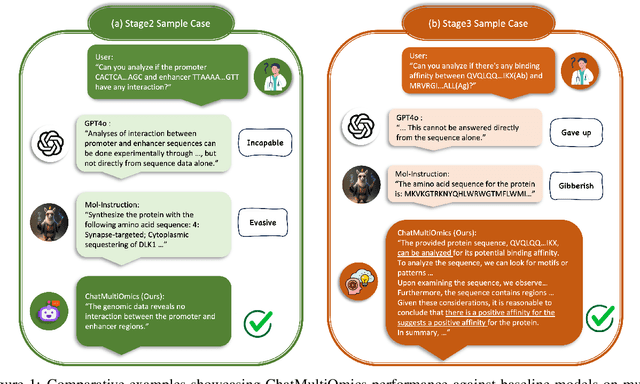
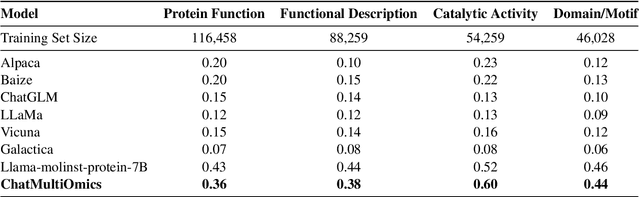
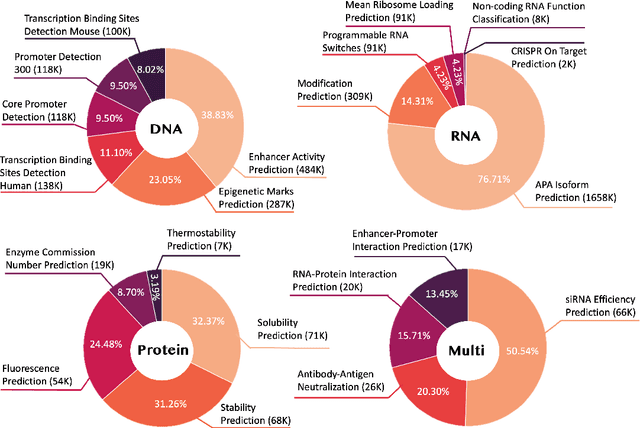
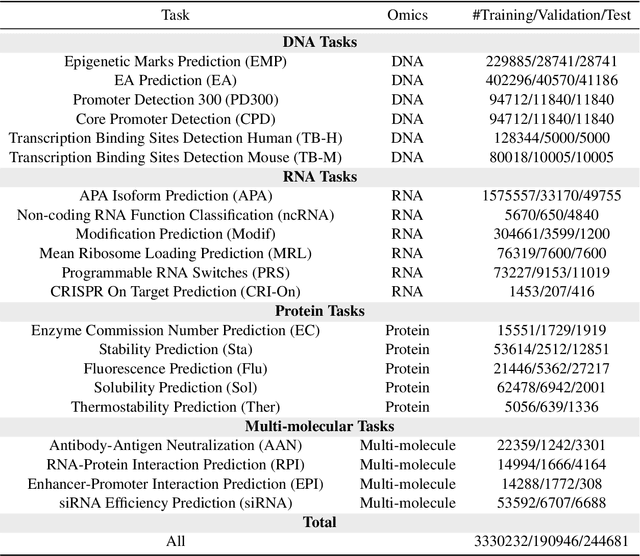
Large language models have already demonstrated their formidable capabilities in general domains, ushering in a revolutionary transformation. However, exploring and exploiting the extensive knowledge of these models to comprehend multi-omics biology remains underexplored. To fill this research gap, we first introduce Biology-Instructions, the first large-scale multi-omics biological sequences-related instruction-tuning dataset including DNA, RNA, proteins, and multi-molecules, designed to bridge the gap between large language models (LLMs) and complex biological sequences-related tasks. This dataset can enhance the versatility of LLMs by integrating diverse biological sequenced-based prediction tasks with advanced reasoning capabilities, while maintaining conversational fluency. Additionally, we reveal significant performance limitations in even state-of-the-art LLMs on biological sequence-related multi-omics tasks without specialized pre-training and instruction-tuning. We further develop a strong baseline called ChatMultiOmics with a novel three-stage training pipeline, demonstrating the powerful ability to understand biology by using Biology-Instructions. Biology-Instructions and ChatMultiOmics are publicly available and crucial resources for enabling more effective integration of LLMs with multi-omics sequence analysis.
COMET: Benchmark for Comprehensive Biological Multi-omics Evaluation Tasks and Language Models
Dec 13, 2024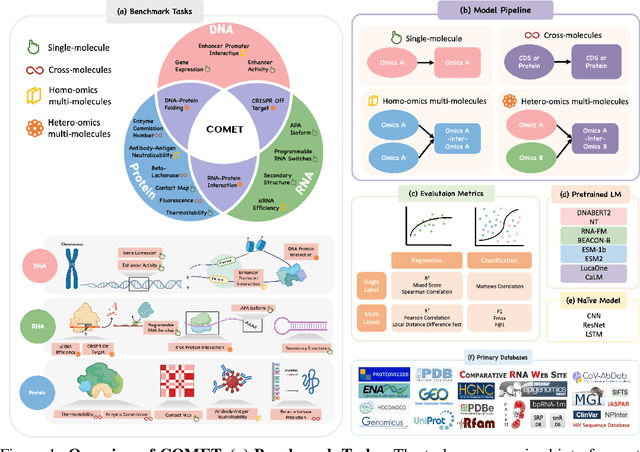
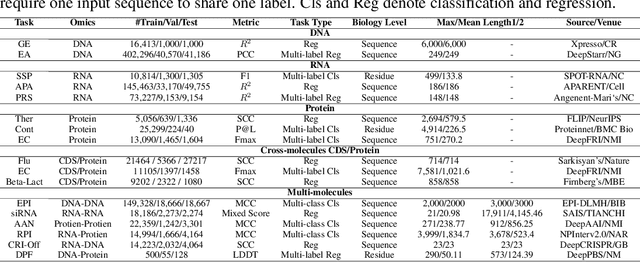
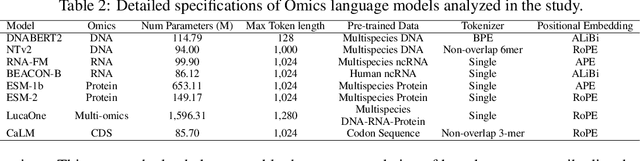
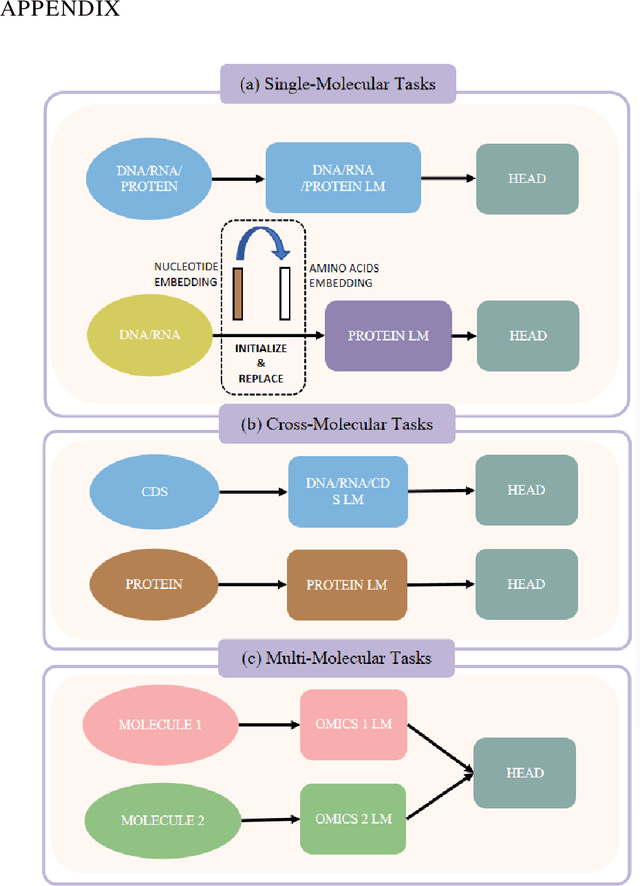
As key elements within the central dogma, DNA, RNA, and proteins play crucial roles in maintaining life by guaranteeing accurate genetic expression and implementation. Although research on these molecules has profoundly impacted fields like medicine, agriculture, and industry, the diversity of machine learning approaches-from traditional statistical methods to deep learning models and large language models-poses challenges for researchers in choosing the most suitable models for specific tasks, especially for cross-omics and multi-omics tasks due to the lack of comprehensive benchmarks. To address this, we introduce the first comprehensive multi-omics benchmark COMET (Benchmark for Biological COmprehensive Multi-omics Evaluation Tasks and Language Models), designed to evaluate models across single-omics, cross-omics, and multi-omics tasks. First, we curate and develop a diverse collection of downstream tasks and datasets covering key structural and functional aspects in DNA, RNA, and proteins, including tasks that span multiple omics levels. Then, we evaluate existing foundational language models for DNA, RNA, and proteins, as well as the newly proposed multi-omics method, offering valuable insights into their performance in integrating and analyzing data from different biological modalities. This benchmark aims to define critical issues in multi-omics research and guide future directions, ultimately promoting advancements in understanding biological processes through integrated and different omics data analysis.
Dynamic PDB: A New Dataset and a SE(3) Model Extension by Integrating Dynamic Behaviors and Physical Properties in Protein Structures
Aug 22, 2024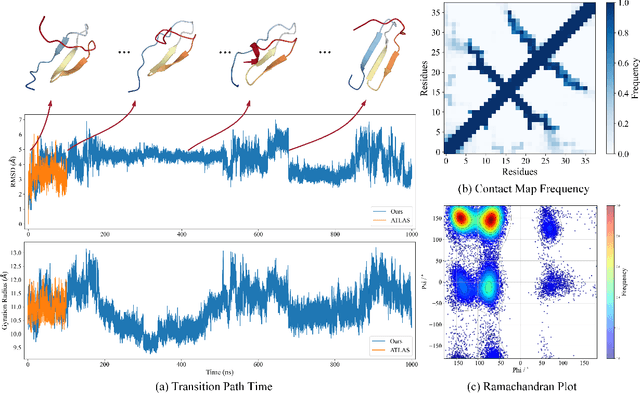
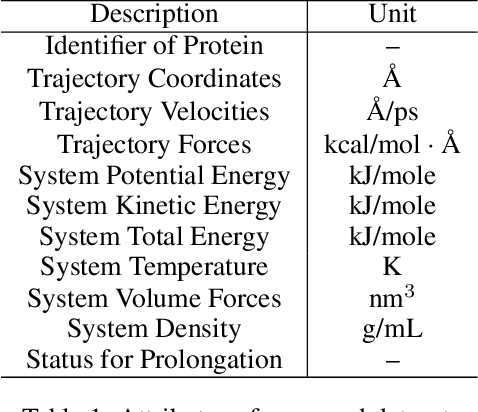
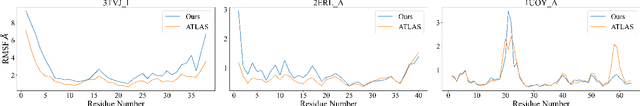
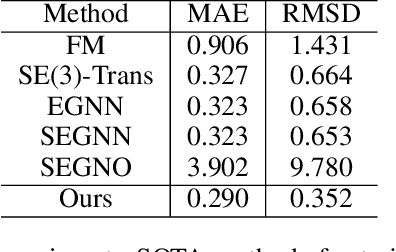
Despite significant progress in static protein structure collection and prediction, the dynamic behavior of proteins, one of their most vital characteristics, has been largely overlooked in prior research. This oversight can be attributed to the limited availability, diversity, and heterogeneity of dynamic protein datasets. To address this gap, we propose to enhance existing prestigious static 3D protein structural databases, such as the Protein Data Bank (PDB), by integrating dynamic data and additional physical properties. Specifically, we introduce a large-scale dataset, Dynamic PDB, encompassing approximately 12.6K proteins, each subjected to all-atom molecular dynamics (MD) simulations lasting 1 microsecond to capture conformational changes. Furthermore, we provide a comprehensive suite of physical properties, including atomic velocities and forces, potential and kinetic energies of proteins, and the temperature of the simulation environment, recorded at 1 picosecond intervals throughout the simulations. For benchmarking purposes, we evaluate state-of-the-art methods on the proposed dataset for the task of trajectory prediction. To demonstrate the value of integrating richer physical properties in the study of protein dynamics and related model design, we base our approach on the SE(3) diffusion model and incorporate these physical properties into the trajectory prediction process. Preliminary results indicate that this straightforward extension of the SE(3) model yields improved accuracy, as measured by MAE and RMSD, when the proposed physical properties are taken into consideration.
4D Diffusion for Dynamic Protein Structure Prediction with Reference Guided Motion Alignment
Aug 22, 2024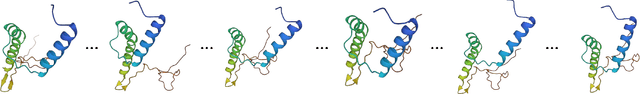
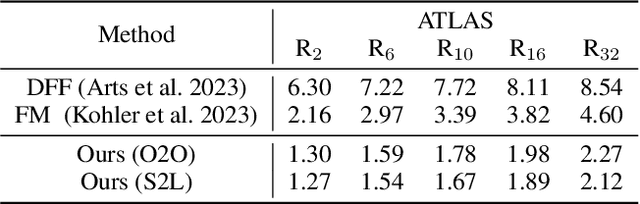
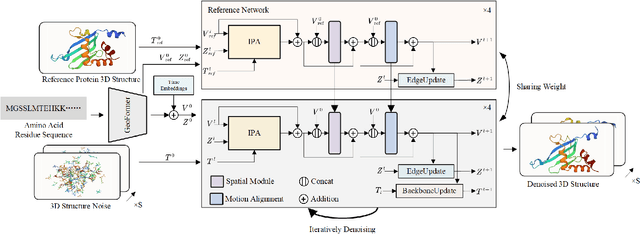
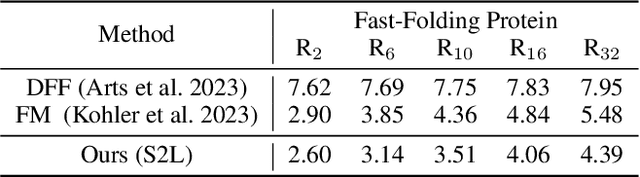
Protein structure prediction is pivotal for understanding the structure-function relationship of proteins, advancing biological research, and facilitating pharmaceutical development and experimental design. While deep learning methods and the expanded availability of experimental 3D protein structures have accelerated structure prediction, the dynamic nature of protein structures has received limited attention. This study introduces an innovative 4D diffusion model incorporating molecular dynamics (MD) simulation data to learn dynamic protein structures. Our approach is distinguished by the following components: (1) a unified diffusion model capable of generating dynamic protein structures, including both the backbone and side chains, utilizing atomic grouping and side-chain dihedral angle predictions; (2) a reference network that enhances structural consistency by integrating the latent embeddings of the initial 3D protein structures; and (3) a motion alignment module aimed at improving temporal structural coherence across multiple time steps. To our knowledge, this is the first diffusion-based model aimed at predicting protein trajectories across multiple time steps simultaneously. Validation on benchmark datasets demonstrates that our model exhibits high accuracy in predicting dynamic 3D structures of proteins containing up to 256 amino acids over 32 time steps, effectively capturing both local flexibility in stable states and significant conformational changes.