 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeSuiren-1.0 Technical Report: A Family of Molecular Foundation Models
Mar 23, 2026We introduce Suiren-1.0, a family of molecular foundation models for the accurate modeling of diverse organic systems. Suiren-1.0 comprising three specialized variants (Suiren-Base, Suiren-Dimer, and Suiren-ConfAvg) is integrated within an algorithmic framework that bridges the gap between 3D conformational geometry and 2D statistical ensemble spaces. We first pre-train Suiren-Base (1.8B parameters) on a 70M-sample Density Functional Theory dataset using spatial self-supervision and SE(3)-equivariant architectures, achieving robust performance in quantum property prediction. Suiren-Dimer extends this capability through continued pre-training on 13.5M intermolecular interaction samples. To enable efficient downstream application, we propose Conformation Compression Distillation (CCD), a diffusion-based framework that distills complex 3D structural representations into 2D conformation-averaged representations. This yields the lightweight Suiren-ConfAvg, which generates high-fidelity representations from SMILES or molecular graphs. Our extensive evaluations demonstrate that Suiren-1.0 establishes state-of-the-art results across a range of tasks. All models and benchmarks are open-sourced.
Equivariant Asynchronous Diffusion: An Adaptive Denoising Schedule for Accelerated Molecular Conformation Generation
Mar 10, 2026Recent 3D molecular generation methods primarily use asynchronous auto-regressive or synchronous diffusion models. While auto-regressive models build molecules sequentially, they're limited by a short horizon and a discrepancy between training and inference. Conversely, synchronous diffusion models denoise all atoms at once, offering a molecule-level horizon but failing to capture the causal relationships inherent in hierarchical molecular structures. We introduce Equivariant Asynchronous Diffusion (EAD) to overcome these limitations. EAD is a novel diffusion model that combines the strengths of both approaches: it uses an asynchronous denoising schedule to better capture molecular hierarchy while maintaining a molecule-level horizon. Since these relationships are often complex, we propose a dynamic scheduling mechanism to adaptively determine the denoising timestep. Experimental results show that EAD achieves state-of-the-art performance in 3D molecular generation.
A Pre-trained Reaction Embedding Descriptor Capturing Bond Transformation Patterns
Jan 07, 2026With the rise of data-driven reaction prediction models, effective reaction descriptors are crucial for bridging the gap between real-world chemistry and digital representations. However, general-purpose, reaction-wise descriptors remain scarce. This study introduces RXNEmb, a novel reaction-level descriptor derived from RXNGraphormer, a model pre-trained to distinguish real reactions from fictitious ones with erroneous bond changes, thereby learning intrinsic bond formation and cleavage patterns. We demonstrate its utility by data-driven re-clustering of the USPTO-50k dataset, yielding a classification that more directly reflects bond-change similarities than rule-based categories. Combined with dimensionality reduction, RXNEmb enables visualization of reaction space diversity. Furthermore, attention weight analysis reveals the model's focus on chemically critical sites, providing mechanistic insight. RXNEmb serves as a powerful, interpretable tool for reaction fingerprinting and analysis, paving the way for more data-centric approaches in reaction analysis and discovery.
Equivariant Spherical Transformer for Efficient Molecular Modeling
May 29, 2025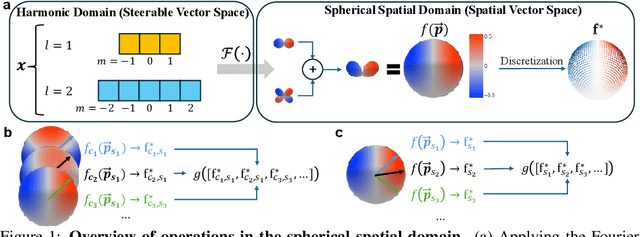
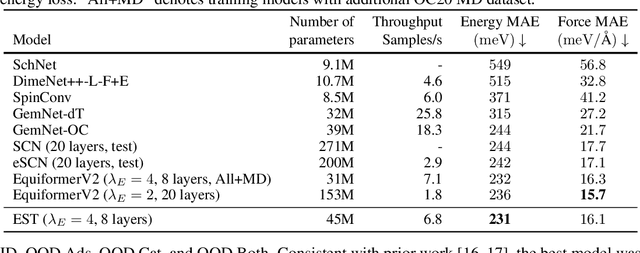
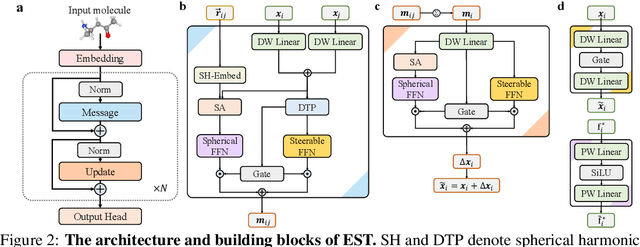
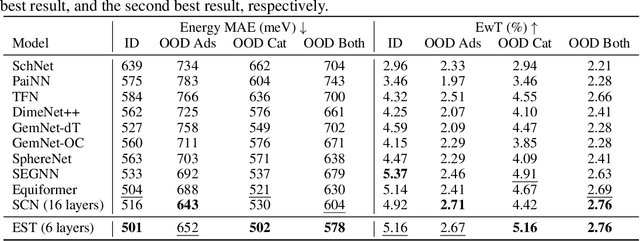
SE(3)-equivariant Graph Neural Networks (GNNs) have significantly advanced molecular system modeling by employing group representations. However, their message passing processes, which rely on tensor product-based convolutions, are limited by insufficient non-linearity and incomplete group representations, thereby restricting expressiveness. To overcome these limitations, we introduce the Equivariant Spherical Transformer (EST), a novel framework that leverages a Transformer structure within the spatial domain of group representations after Fourier transform. We theoretically and empirically demonstrate that EST can encompass the function space of tensor products while achieving superior expressiveness. Furthermore, EST's equivariant inductive bias is guaranteed through a uniform sampling strategy for the Fourier transform. Our experiments demonstrate state-of-the-art performance by EST on various molecular benchmarks, including OC20 and QM9.
ChemHAS: Hierarchical Agent Stacking for Enhancing Chemistry Tools
May 27, 2025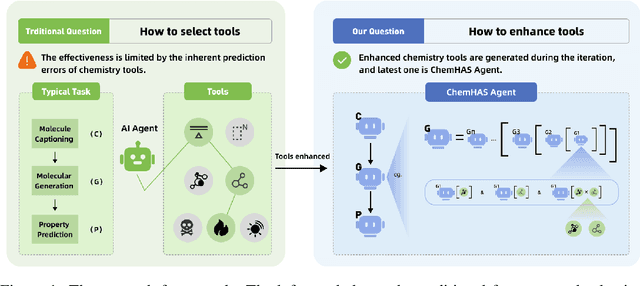
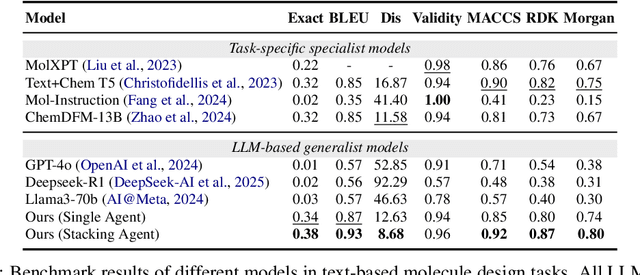
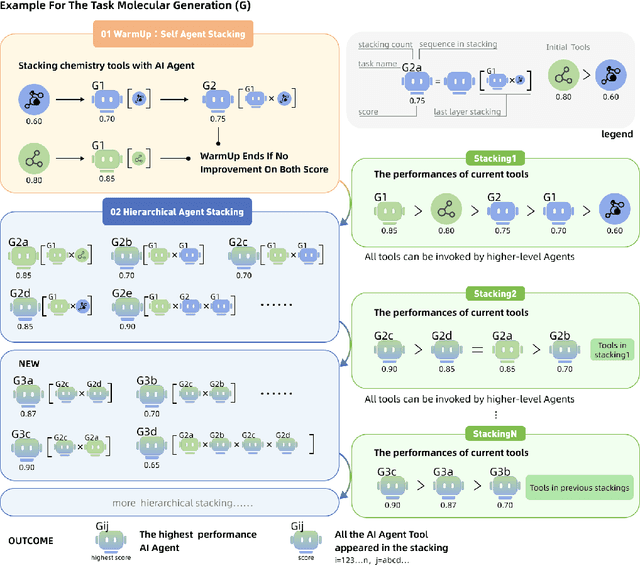
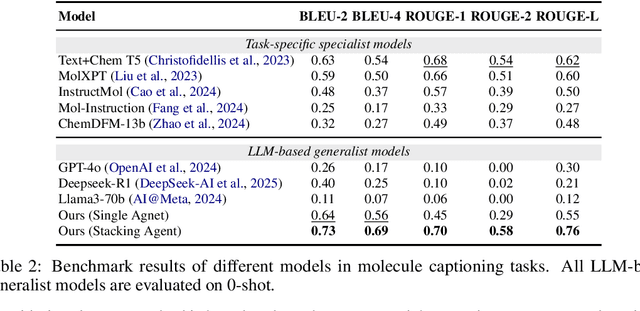
Large Language Model (LLM)-based agents have demonstrated the ability to improve performance in chemistry-related tasks by selecting appropriate tools. However, their effectiveness remains limited by the inherent prediction errors of chemistry tools. In this paper, we take a step further by exploring how LLMbased agents can, in turn, be leveraged to reduce prediction errors of the tools. To this end, we propose ChemHAS (Chemical Hierarchical Agent Stacking), a simple yet effective method that enhances chemistry tools through optimizing agent-stacking structures from limited data. ChemHAS achieves state-of-the-art performance across four fundamental chemistry tasks, demonstrating that our method can effectively compensate for prediction errors of the tools. Furthermore, we identify and characterize four distinct agent-stacking behaviors, potentially improving interpretability and revealing new possibilities for AI agent applications in scientific research. Our code and dataset are publicly available at https: //anonymous.4open.science/r/ChemHAS-01E4/README.md.
Equivariant Masked Position Prediction for Efficient Molecular Representation
Feb 12, 2025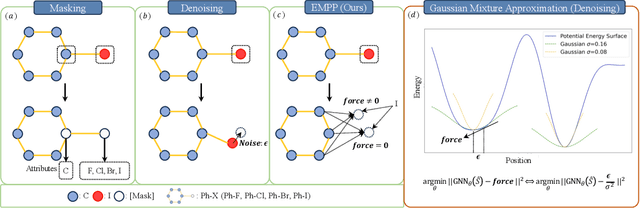
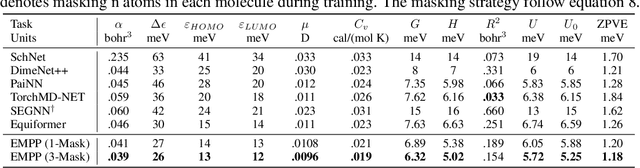
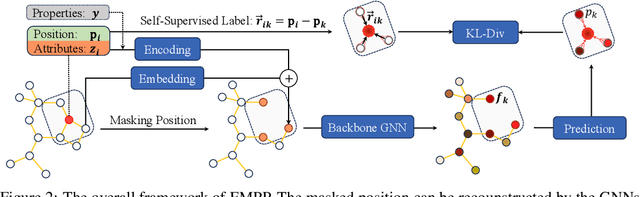
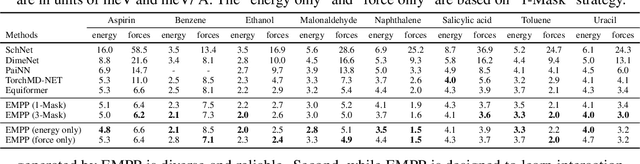
Graph neural networks (GNNs) have shown considerable promise in computational chemistry. However, the limited availability of molecular data raises concerns regarding GNNs' ability to effectively capture the fundamental principles of physics and chemistry, which constrains their generalization capabilities. To address this challenge, we introduce a novel self-supervised approach termed Equivariant Masked Position Prediction (EMPP), grounded in intramolecular potential and force theory. Unlike conventional attribute masking techniques, EMPP formulates a nuanced position prediction task that is more well-defined and enhances the learning of quantum mechanical features. EMPP also bypasses the approximation of the Gaussian mixture distribution commonly used in denoising methods, allowing for more accurate acquisition of physical properties. Experimental results indicate that EMPP significantly enhances performance of advanced molecular architectures, surpassing state-of-the-art self-supervised approaches. Our code is released in https://github.com/ajy112/EMPP.
* 24 pages, 6 figures
Dynamic PDB: A New Dataset and a SE(3) Model Extension by Integrating Dynamic Behaviors and Physical Properties in Protein Structures
Aug 22, 2024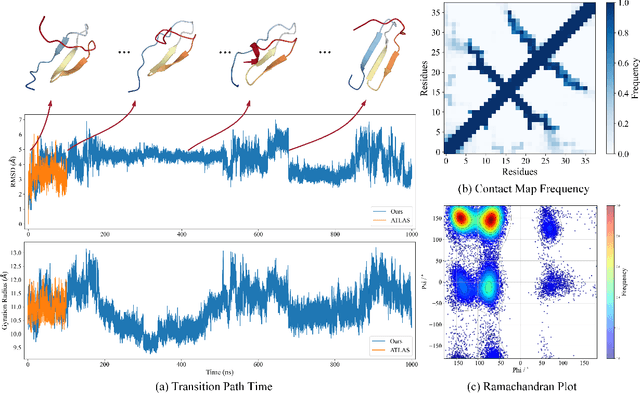
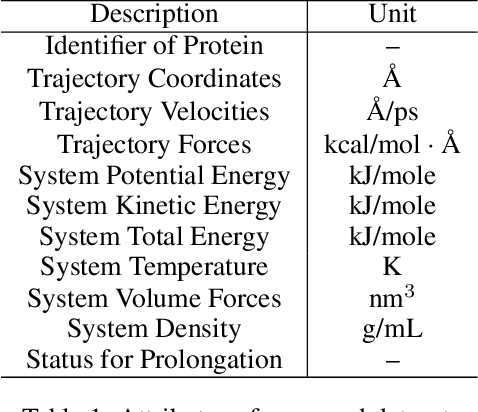
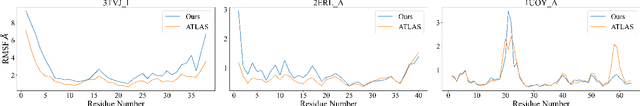
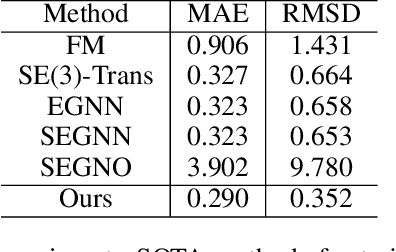
Despite significant progress in static protein structure collection and prediction, the dynamic behavior of proteins, one of their most vital characteristics, has been largely overlooked in prior research. This oversight can be attributed to the limited availability, diversity, and heterogeneity of dynamic protein datasets. To address this gap, we propose to enhance existing prestigious static 3D protein structural databases, such as the Protein Data Bank (PDB), by integrating dynamic data and additional physical properties. Specifically, we introduce a large-scale dataset, Dynamic PDB, encompassing approximately 12.6K proteins, each subjected to all-atom molecular dynamics (MD) simulations lasting 1 microsecond to capture conformational changes. Furthermore, we provide a comprehensive suite of physical properties, including atomic velocities and forces, potential and kinetic energies of proteins, and the temperature of the simulation environment, recorded at 1 picosecond intervals throughout the simulations. For benchmarking purposes, we evaluate state-of-the-art methods on the proposed dataset for the task of trajectory prediction. To demonstrate the value of integrating richer physical properties in the study of protein dynamics and related model design, we base our approach on the SE(3) diffusion model and incorporate these physical properties into the trajectory prediction process. Preliminary results indicate that this straightforward extension of the SE(3) model yields improved accuracy, as measured by MAE and RMSD, when the proposed physical properties are taken into consideration.