 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeRank-and-Reason: Multi-Agent Collaboration Accelerates Zero-Shot Protein Mutation Prediction
Feb 03, 2026Zero-shot mutation prediction is vital for low-resource protein engineering, yet existing protein language models (PLMs) often yield statistically confident results that ignore fundamental biophysical constraints. Currently, selecting candidates for wet-lab validation relies on manual expert auditing of PLM outputs, a process that is inefficient, subjective, and highly dependent on domain expertise. To address this, we propose Rank-and-Reason (VenusRAR), a two-stage agentic framework to automate this workflow and maximize expected wet-lab fitness. In the Rank-Stage, a Computational Expert and Virtual Biologist aggregate a context-aware multi-modal ensemble, establishing a new Spearman correlation record of 0.551 (vs. 0.518) on ProteinGym. In the Reason-Stage, an agentic Expert Panel employs chain-of-thought reasoning to audit candidates against geometric and structural constraints, improving the Top-5 Hit Rate by up to 367% on ProteinGym-DMS99. The wet-lab validation on Cas12i3 nuclease further confirms the framework's efficacy, achieving a 46.7% positive rate and identifying two novel mutants with 4.23-fold and 5.05-fold activity improvements. Code and datasets are released on GitHub (https://github.com/ai4protein/VenusRAR/).
VenusFactory: A Unified Platform for Protein Engineering Data Retrieval and Language Model Fine-Tuning
Mar 19, 2025Natural language processing (NLP) has significantly influenced scientific domains beyond human language, including protein engineering, where pre-trained protein language models (PLMs) have demonstrated remarkable success. However, interdisciplinary adoption remains limited due to challenges in data collection, task benchmarking, and application. This work presents VenusFactory, a versatile engine that integrates biological data retrieval, standardized task benchmarking, and modular fine-tuning of PLMs. VenusFactory supports both computer science and biology communities with choices of both a command-line execution and a Gradio-based no-code interface, integrating $40+$ protein-related datasets and $40+$ popular PLMs. All implementations are open-sourced on https://github.com/tyang816/VenusFactory.
CPE-Pro: A Structure-Sensitive Deep Learning Model for Protein Representation and Origin Evaluation
Oct 21, 2024

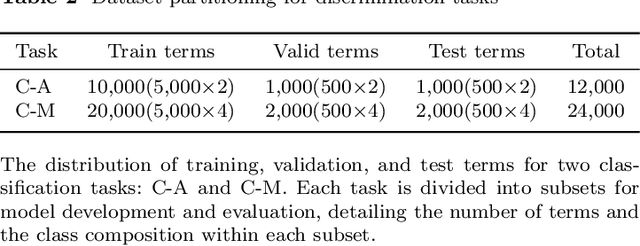

Protein structures are important for understanding their functions and interactions. Currently, many protein structure prediction methods are enriching the structure database. Discriminating the origin of structures is crucial for distinguishing between experimentally resolved and computationally predicted structures, evaluating the reliability of prediction methods, and guiding downstream biological studies. Building on works in structure prediction, We developed a structure-sensitive supervised deep learning model, Crystal vs Predicted Evaluator for Protein Structure (CPE-Pro), to represent and discriminate the origin of protein structures. CPE-Pro learns the structural information of proteins and captures inter-structural differences to achieve accurate traceability on four data classes, and is expected to be extended to more. Simultaneously, we utilized Foldseek to encode protein structures into "structure-sequence" and trained a protein Structural Sequence Language Model, SSLM. Preliminary experiments demonstrated that, compared to large-scale protein language models pre-trained on vast amounts of amino acid sequences, the "structure-sequences" enable the language model to learn more informative protein features, enhancing and optimizing structural representations. We have provided the code, model weights, and all related materials on https://github.com/GouWenrui/CPE-Pro-main.git.
A PLMs based protein retrieval framework
Jul 16, 2024



Protein retrieval, which targets the deconstruction of the relationship between sequences, structures and functions, empowers the advancing of biology. Basic Local Alignment Search Tool (BLAST), a sequence-similarity-based algorithm, has proved the efficiency of this field. Despite the existing tools for protein retrieval, they prioritize sequence similarity and probably overlook proteins that are dissimilar but share homology or functionality. In order to tackle this problem, we propose a novel protein retrieval framework that mitigates the bias towards sequence similarity. Our framework initiatively harnesses protein language models (PLMs) to embed protein sequences within a high-dimensional feature space, thereby enhancing the representation capacity for subsequent analysis. Subsequently, an accelerated indexed vector database is constructed to facilitate expedited access and retrieval of dense vectors. Extensive experiments demonstrate that our framework can equally retrieve both similar and dissimilar proteins. Moreover, this approach enables the identification of proteins that conventional methods fail to uncover. This framework will effectively assist in protein mining and empower the development of biology.
Simple, Efficient and Scalable Structure-aware Adapter Boosts Protein Language Models
Apr 23, 2024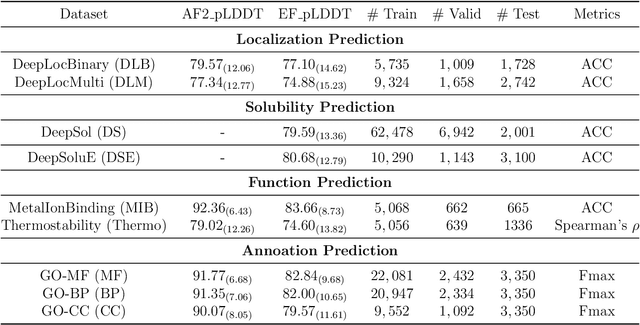
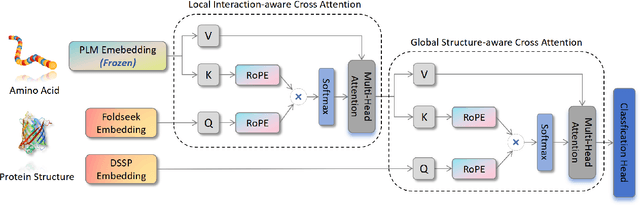
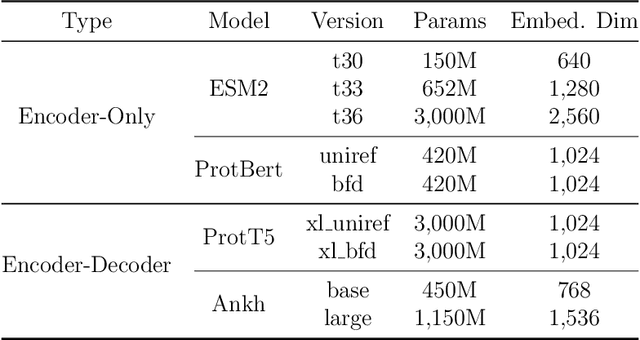
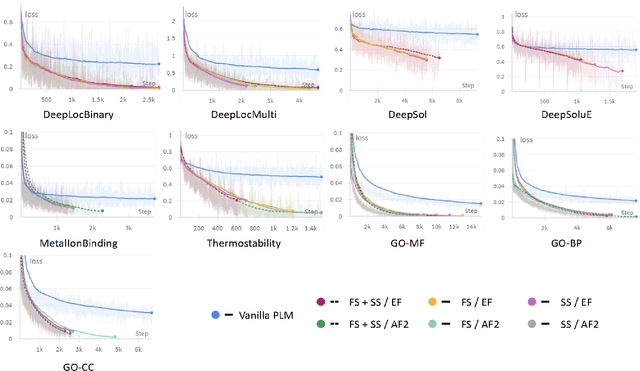
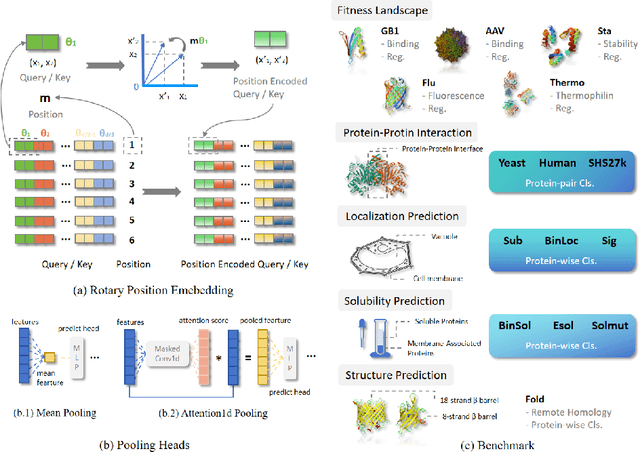
Fine-tuning Pre-trained protein language models (PLMs) has emerged as a prominent strategy for enhancing downstream prediction tasks, often outperforming traditional supervised learning approaches. As a widely applied powerful technique in natural language processing, employing Parameter-Efficient Fine-Tuning techniques could potentially enhance the performance of PLMs. However, the direct transfer to life science tasks is non-trivial due to the different training strategies and data forms. To address this gap, we introduce SES-Adapter, a simple, efficient, and scalable adapter method for enhancing the representation learning of PLMs. SES-Adapter incorporates PLM embeddings with structural sequence embeddings to create structure-aware representations. We show that the proposed method is compatible with different PLM architectures and across diverse tasks. Extensive evaluations are conducted on 2 types of folding structures with notable quality differences, 9 state-of-the-art baselines, and 9 benchmark datasets across distinct downstream tasks. Results show that compared to vanilla PLMs, SES-Adapter improves downstream task performance by a maximum of 11% and an average of 3%, with significantly accelerated training speed by a maximum of 1034% and an average of 362%, the convergence rate is also improved by approximately 2 times. Moreover, positive optimization is observed even with low-quality predicted structures. The source code for SES-Adapter is available at https://github.com/tyang816/SES-Adapter.
PETA: Evaluating the Impact of Protein Transfer Learning with Sub-word Tokenization on Downstream Applications
Oct 26, 2023

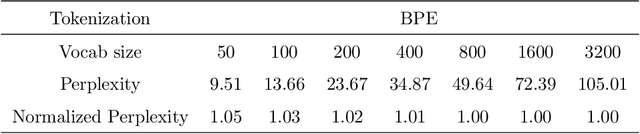
Large protein language models are adept at capturing the underlying evolutionary information in primary structures, offering significant practical value for protein engineering. Compared to natural language models, protein amino acid sequences have a smaller data volume and a limited combinatorial space. Choosing an appropriate vocabulary size to optimize the pre-trained model is a pivotal issue. Moreover, despite the wealth of benchmarks and studies in the natural language community, there remains a lack of a comprehensive benchmark for systematically evaluating protein language model quality. Given these challenges, PETA trained language models with 14 different vocabulary sizes under three tokenization methods. It conducted thousands of tests on 33 diverse downstream datasets to assess the models' transfer learning capabilities, incorporating two classification heads and three random seeds to mitigate potential biases. Extensive experiments indicate that vocabulary sizes between 50 and 200 optimize the model, whereas sizes exceeding 800 detrimentally affect the model's representational performance. Our code, model weights and datasets are available at https://github.com/ginnm/ProteinPretraining.
MedChatZH: a Better Medical Adviser Learns from Better Instructions
Sep 03, 2023


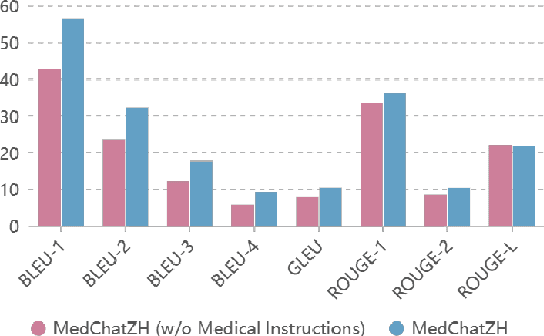
Generative large language models (LLMs) have shown great success in various applications, including question-answering (QA) and dialogue systems. However, in specialized domains like traditional Chinese medical QA, these models may perform unsatisfactorily without fine-tuning on domain-specific datasets. To address this, we introduce MedChatZH, a dialogue model designed specifically for traditional Chinese medical QA. Our model is pre-trained on Chinese traditional medical books and fine-tuned with a carefully curated medical instruction dataset. It outperforms several solid baselines on a real-world medical dialogue dataset. We release our model, code, and dataset on https://github.com/tyang816/MedChatZH to facilitate further research in the domain of traditional Chinese medicine and LLMs.
SESNet: sequence-structure feature-integrated deep learning method for data-efficient protein engineering
Dec 29, 2022Deep learning has been widely used for protein engineering. However, it is limited by the lack of sufficient experimental data to train an accurate model for predicting the functional fitness of high-order mutants. Here, we develop SESNet, a supervised deep-learning model to predict the fitness for protein mutants by leveraging both sequence and structure information, and exploiting attention mechanism. Our model integrates local evolutionary context from homologous sequences, the global evolutionary context encoding rich semantic from the universal protein sequence space and the structure information accounting for the microenvironment around each residue in a protein. We show that SESNet outperforms state-of-the-art models for predicting the sequence-function relationship on 26 deep mutational scanning datasets. More importantly, we propose a data augmentation strategy by leveraging the data from unsupervised models to pre-train our model. After that, our model can achieve strikingly high accuracy in prediction of the fitness of protein mutants, especially for the higher order variants (> 4 mutation sites), when finetuned by using only a small number of experimental mutation data (<50). The strategy proposed is of great practical value as the required experimental effort, i.e., producing a few tens of experimental mutation data on a given protein, is generally affordable by an ordinary biochemical group and can be applied on almost any protein.