 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeShapley-Guided Neural Repair Approach via Derivative-Free Optimization
Apr 01, 2026DNNs are susceptible to defects like backdoors, adversarial attacks, and unfairness, undermining their reliability. Existing approaches mainly involve retraining, optimization, constraint-solving, or search algorithms. However, most methods rely on gradient calculations, restricting applicability to specific activation functions (e.g., ReLU), or use search algorithms with uninterpretable localization and repair. Furthermore, they often lack generalizability across multiple properties. We propose SHARPEN, integrating interpretable fault localization with a derivative-free optimization strategy. First, SHARPEN introduces a Deep SHAP-based localization strategy quantifying each layer's and neuron's marginal contribution to erroneous outputs. Specifically, a hierarchical coarse-to-fine approach reranks layers by aggregated impact, then locates faulty neurons/filters by analyzing activation divergences between property-violating and benign states. Subsequently, SHARPEN incorporates CMA-ES to repair identified neurons. CMA-ES leverages a covariance matrix to capture variable dependencies, enabling gradient-free search and coordinated adjustments across coupled neurons. By combining interpretable localization with evolutionary optimization, SHARPEN enables derivative-free repair across architectures, being less sensitive to gradient anomalies and hyperparameters. We demonstrate SHARPEN's effectiveness on three repair tasks. Balancing property repair and accuracy preservation, it outperforms baselines in backdoor removal (+10.56%), adversarial mitigation (+5.78%), and unfairness repair (+11.82%). Notably, SHARPEN handles diverse tasks, and its modular design is plug-and-play with different derivative-free optimizers, highlighting its flexibility.
Self-evolving AI agents for protein discovery and directed evolution
Mar 28, 2026Protein scientific discovery is bottlenecked by the manual orchestration of information and algorithms, while general agents are insufficient in complex domain projects. VenusFactory2 provides an autonomous framework that shifts from static tool usage to dynamic workflow synthesis via a self-evolving multi-agent infrastructure to address protein-related demands. It outperforms a set of well-known agents on the VenusAgentEval benchmark, and autonomously organizes the discovery and optimization of proteins from a single natural language prompt.
Rank-and-Reason: Multi-Agent Collaboration Accelerates Zero-Shot Protein Mutation Prediction
Feb 03, 2026Zero-shot mutation prediction is vital for low-resource protein engineering, yet existing protein language models (PLMs) often yield statistically confident results that ignore fundamental biophysical constraints. Currently, selecting candidates for wet-lab validation relies on manual expert auditing of PLM outputs, a process that is inefficient, subjective, and highly dependent on domain expertise. To address this, we propose Rank-and-Reason (VenusRAR), a two-stage agentic framework to automate this workflow and maximize expected wet-lab fitness. In the Rank-Stage, a Computational Expert and Virtual Biologist aggregate a context-aware multi-modal ensemble, establishing a new Spearman correlation record of 0.551 (vs. 0.518) on ProteinGym. In the Reason-Stage, an agentic Expert Panel employs chain-of-thought reasoning to audit candidates against geometric and structural constraints, improving the Top-5 Hit Rate by up to 367% on ProteinGym-DMS99. The wet-lab validation on Cas12i3 nuclease further confirms the framework's efficacy, achieving a 46.7% positive rate and identifying two novel mutants with 4.23-fold and 5.05-fold activity improvements. Code and datasets are released on GitHub (https://github.com/ai4protein/VenusRAR/).
Probabilistic Temporal Masked Attention for Cross-view Online Action Detection
Aug 23, 2025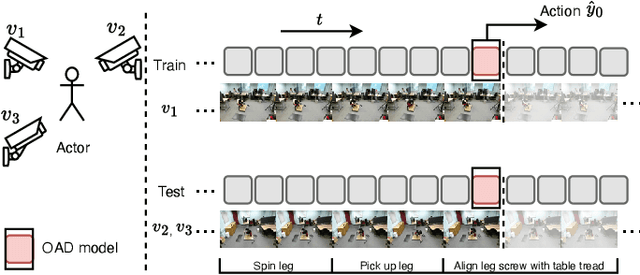
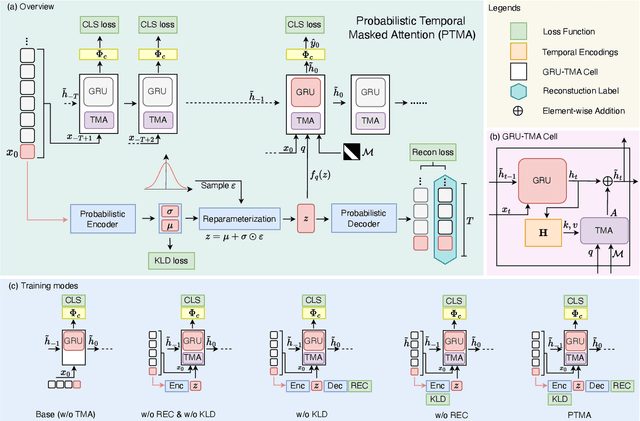
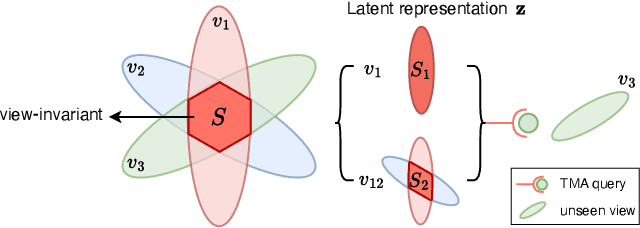
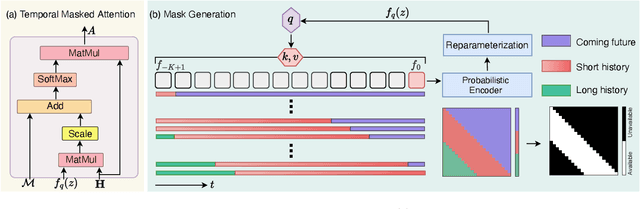
As a critical task in video sequence classification within computer vision, Online Action Detection (OAD) has garnered significant attention. The sensitivity of mainstream OAD models to varying video viewpoints often hampers their generalization when confronted with unseen sources. To address this limitation, we propose a novel Probabilistic Temporal Masked Attention (PTMA) model, which leverages probabilistic modeling to derive latent compressed representations of video frames in a cross-view setting. The PTMA model incorporates a GRU-based temporal masked attention (TMA) cell, which leverages these representations to effectively query the input video sequence, thereby enhancing information interaction and facilitating autoregressive frame-level video analysis. Additionally, multi-view information can be integrated into the probabilistic modeling to facilitate the extraction of view-invariant features. Experiments conducted under three evaluation protocols: cross-subject (cs), cross-view (cv), and cross-subject-view (csv) show that PTMA achieves state-of-the-art performance on the DAHLIA, IKEA ASM, and Breakfast datasets.
VenusX: Unlocking Fine-Grained Functional Understanding of Proteins
May 17, 2025



Deep learning models have driven significant progress in predicting protein function and interactions at the protein level. While these advancements have been invaluable for many biological applications such as enzyme engineering and function annotation, a more detailed perspective is essential for understanding protein functional mechanisms and evaluating the biological knowledge captured by models. To address this demand, we introduce VenusX, the first large-scale benchmark for fine-grained functional annotation and function-based protein pairing at the residue, fragment, and domain levels. VenusX comprises three major task categories across six types of annotations, including residue-level binary classification, fragment-level multi-class classification, and pairwise functional similarity scoring for identifying critical active sites, binding sites, conserved sites, motifs, domains, and epitopes. The benchmark features over 878,000 samples curated from major open-source databases such as InterPro, BioLiP, and SAbDab. By providing mixed-family and cross-family splits at three sequence identity thresholds, our benchmark enables a comprehensive assessment of model performance on both in-distribution and out-of-distribution scenarios. For baseline evaluation, we assess a diverse set of popular and open-source models, including pre-trained protein language models, sequence-structure hybrids, structure-based methods, and alignment-based techniques. Their performance is reported across all benchmark datasets and evaluation settings using multiple metrics, offering a thorough comparison and a strong foundation for future research. Code and data are publicly available at https://github.com/ai4protein/VenusX.
Transferable Mask Transformer: Cross-domain Semantic Segmentation with Region-adaptive Transferability Estimation
Apr 08, 2025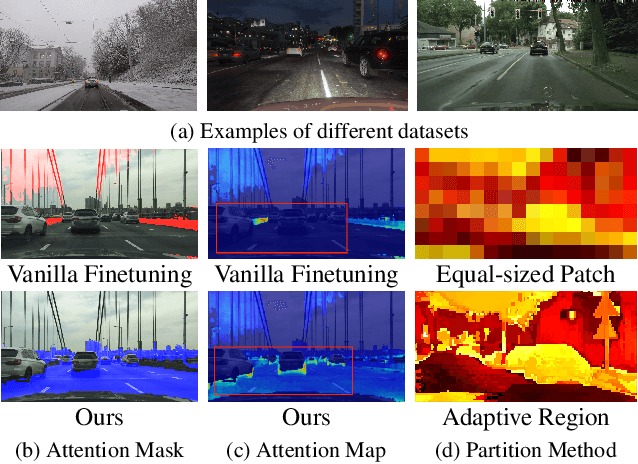
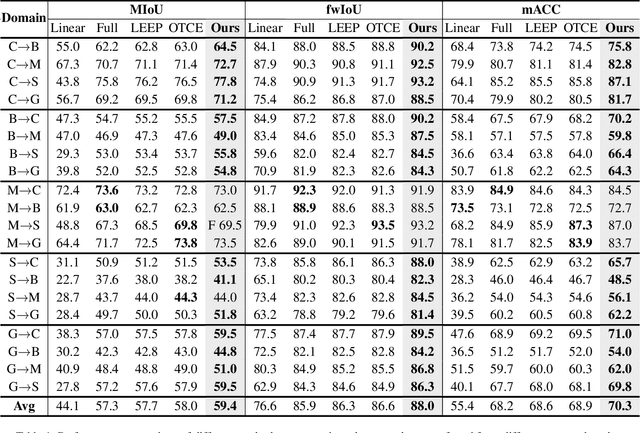
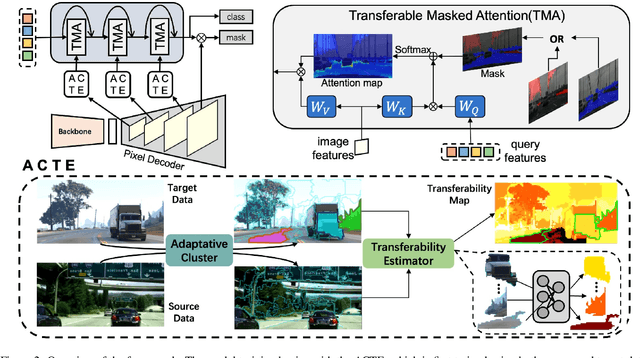
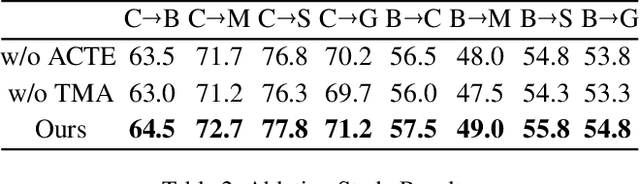
Recent advances in Vision Transformers (ViTs) have set new benchmarks in semantic segmentation. However, when adapting pretrained ViTs to new target domains, significant performance degradation often occurs due to distribution shifts, resulting in suboptimal global attention. Since self-attention mechanisms are inherently data-driven, they may fail to effectively attend to key objects when source and target domains exhibit differences in texture, scale, or object co-occurrence patterns. While global and patch-level domain adaptation methods provide partial solutions, region-level adaptation with dynamically shaped regions is crucial due to spatial heterogeneity in transferability across different image areas. We present Transferable Mask Transformer (TMT), a novel region-level adaptation framework for semantic segmentation that aligns cross-domain representations through spatial transferability analysis. TMT consists of two key components: (1) An Adaptive Cluster-based Transferability Estimator (ACTE) that dynamically segments images into structurally and semantically coherent regions for localized transferability assessment, and (2) A Transferable Masked Attention (TMA) module that integrates region-specific transferability maps into ViTs' attention mechanisms, prioritizing adaptation in regions with low transferability and high semantic uncertainty. Comprehensive evaluations across 20 cross-domain pairs demonstrate TMT's superiority, achieving an average 2% MIoU improvement over vanilla fine-tuning and a 1.28% increase compared to state-of-the-art baselines. The source code will be publicly available.
VenusFactory: A Unified Platform for Protein Engineering Data Retrieval and Language Model Fine-Tuning
Mar 19, 2025Natural language processing (NLP) has significantly influenced scientific domains beyond human language, including protein engineering, where pre-trained protein language models (PLMs) have demonstrated remarkable success. However, interdisciplinary adoption remains limited due to challenges in data collection, task benchmarking, and application. This work presents VenusFactory, a versatile engine that integrates biological data retrieval, standardized task benchmarking, and modular fine-tuning of PLMs. VenusFactory supports both computer science and biology communities with choices of both a command-line execution and a Gradio-based no-code interface, integrating $40+$ protein-related datasets and $40+$ popular PLMs. All implementations are open-sourced on https://github.com/tyang816/VenusFactory.
Transfer Risk Map: Mitigating Pixel-level Negative Transfer in Medical Segmentation
Feb 04, 2025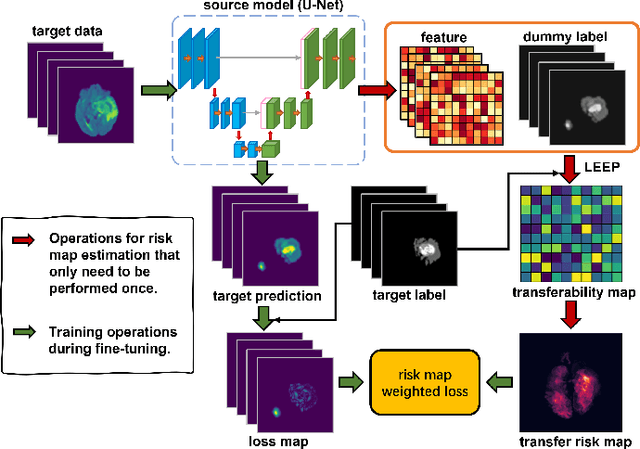



How to mitigate negative transfer in transfer learning is a long-standing and challenging issue, especially in the application of medical image segmentation. Existing methods for reducing negative transfer focus on classification or regression tasks, ignoring the non-uniform negative transfer risk in different image regions. In this work, we propose a simple yet effective weighted fine-tuning method that directs the model's attention towards regions with significant transfer risk for medical semantic segmentation. Specifically, we compute a transferability-guided transfer risk map to quantify the transfer hardness for each pixel and the potential risks of negative transfer. During the fine-tuning phase, we introduce a map-weighted loss function, normalized with image foreground size to counter class imbalance. Extensive experiments on brain segmentation datasets show our method significantly improves the target task performance, with gains of 4.37% on FeTS2021 and 1.81% on iSeg2019, avoiding negative transfer across modalities and tasks. Meanwhile, a 2.9% gain under a few-shot scenario validates the robustness of our approach.
Retrieval-Enhanced Mutation Mastery: Augmenting Zero-Shot Prediction of Protein Language Model
Oct 28, 2024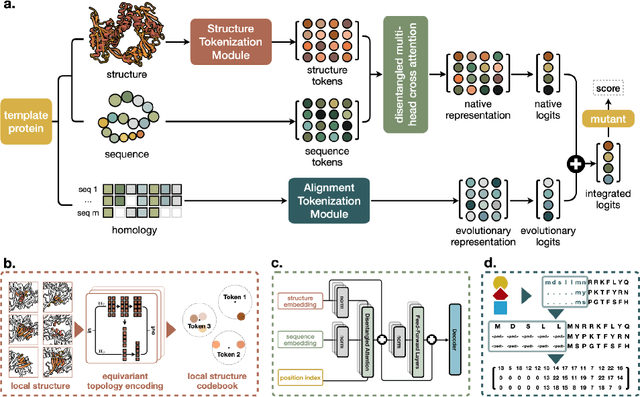
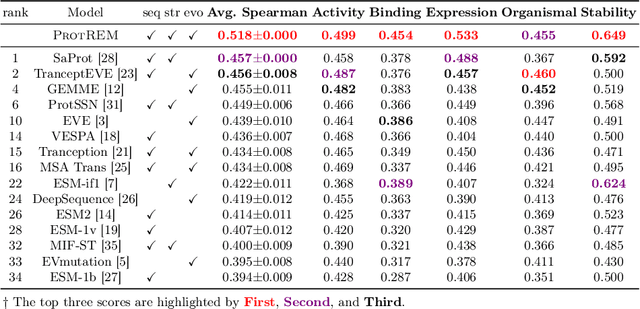
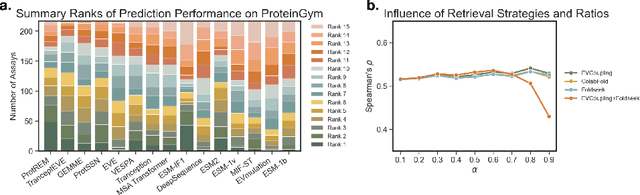
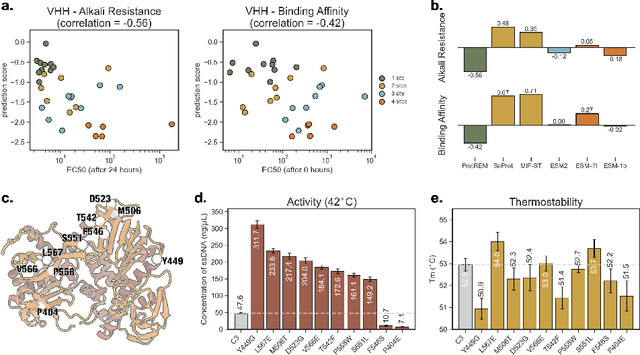
Enzyme engineering enables the modification of wild-type proteins to meet industrial and research demands by enhancing catalytic activity, stability, binding affinities, and other properties. The emergence of deep learning methods for protein modeling has demonstrated superior results at lower costs compared to traditional approaches such as directed evolution and rational design. In mutation effect prediction, the key to pre-training deep learning models lies in accurately interpreting the complex relationships among protein sequence, structure, and function. This study introduces a retrieval-enhanced protein language model for comprehensive analysis of native properties from sequence and local structural interactions, as well as evolutionary properties from retrieved homologous sequences. The state-of-the-art performance of the proposed ProtREM is validated on over 2 million mutants across 217 assays from an open benchmark (ProteinGym). We also conducted post-hoc analyses of the model's ability to improve the stability and binding affinity of a VHH antibody. Additionally, we designed 10 new mutants on a DNA polymerase and conducted wet-lab experiments to evaluate their enhanced activity at higher temperatures. Both in silico and experimental evaluations confirmed that our method provides reliable predictions of mutation effects, offering an auxiliary tool for biologists aiming to evolve existing enzymes. The implementation is publicly available at https://github.com/tyang816/ProtREM.
Immunogenicity Prediction with Dual Attention Enables Vaccine Target Selection
Oct 03, 2024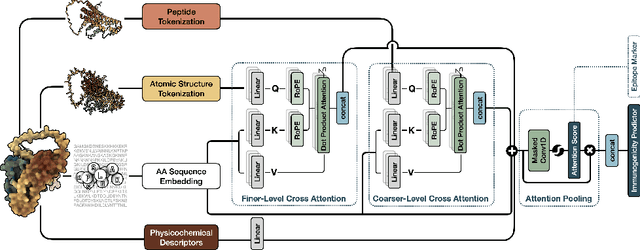
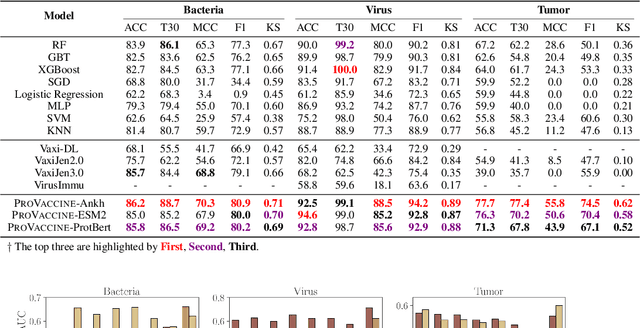
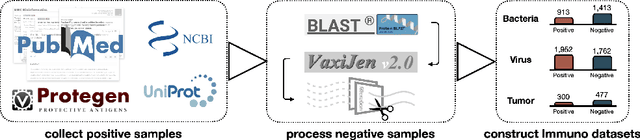

Immunogenicity prediction is a central topic in reverse vaccinology for finding candidate vaccines that can trigger protective immune responses. Existing approaches typically rely on highly compressed features and simple model architectures, leading to limited prediction accuracy and poor generalizability. To address these challenges, we introduce ProVaccine, a novel deep learning solution with a dual attention mechanism that integrates pre-trained latent vector representations of protein sequences and structures. We also compile the most comprehensive immunogenicity dataset to date, encompassing over 9,500 antigen sequences, structures, and immunogenicity labels from bacteria, viruses, and tumors. Extensive experiments demonstrate that ProVaccine outperforms existing methods across a wide range of evaluation metrics. Furthermore, we establish a post-hoc validation protocol to assess the practical significance of deep learning models in tackling vaccine design challenges. Our work provides an effective tool for vaccine design and sets valuable benchmarks for future research.