 Add to Chrome
Add to Chrome Add to Firefox
Add to Firefox Add to Edge
Add to EdgeCell-ontology guided transcriptome foundation model
Aug 22, 2024



Transcriptome foundation models TFMs hold great promises of deciphering the transcriptomic language that dictate diverse cell functions by self-supervised learning on large-scale single-cell gene expression data, and ultimately unraveling the complex mechanisms of human diseases. However, current TFMs treat cells as independent samples and ignore the taxonomic relationships between cell types, which are available in cell ontology graphs. We argue that effectively leveraging this ontology information during the TFM pre-training can improve learning biologically meaningful gene co-expression patterns while preserving TFM as a general purpose foundation model for downstream zero-shot and fine-tuning tasks. To this end, we present \textbf{s}ingle \textbf{c}ell, \textbf{Cell}-\textbf{o}ntology guided TFM scCello. We introduce cell-type coherence loss and ontology alignment loss, which are minimized along with the masked gene expression prediction loss during the pre-training. The novel loss component guide scCello to learn the cell-type-specific representation and the structural relation between cell types from the cell ontology graph, respectively. We pre-trained scCello on 22 million cells from CellxGene database leveraging their cell-type labels mapped to the cell ontology graph from Open Biological and Biomedical Ontology Foundry. Our TFM demonstrates competitive generalization and transferability performance over the existing TFMs on biologically important tasks including identifying novel cell types of unseen cells, prediction of cell-type-specific marker genes, and cancer drug responses.
Evaluating representation learning on the protein structure universe
Jun 19, 2024We introduce ProteinWorkshop, a comprehensive benchmark suite for representation learning on protein structures with Geometric Graph Neural Networks. We consider large-scale pre-training and downstream tasks on both experimental and predicted structures to enable the systematic evaluation of the quality of the learned structural representation and their usefulness in capturing functional relationships for downstream tasks. We find that: (1) large-scale pretraining on AlphaFold structures and auxiliary tasks consistently improve the performance of both rotation-invariant and equivariant GNNs, and (2) more expressive equivariant GNNs benefit from pretraining to a greater extent compared to invariant models. We aim to establish a common ground for the machine learning and computational biology communities to rigorously compare and advance protein structure representation learning. Our open-source codebase reduces the barrier to entry for working with large protein structure datasets by providing: (1) storage-efficient dataloaders for large-scale structural databases including AlphaFoldDB and ESM Atlas, as well as (2) utilities for constructing new tasks from the entire PDB. ProteinWorkshop is available at: github.com/a-r-j/ProteinWorkshop.
Fusing Neural and Physical: Augment Protein Conformation Sampling with Tractable Simulations
Feb 16, 2024The protein dynamics are common and important for their biological functions and properties, the study of which usually involves time-consuming molecular dynamics (MD) simulations in silico. Recently, generative models has been leveraged as a surrogate sampler to obtain conformation ensembles with orders of magnitude faster and without requiring any simulation data (a "zero-shot" inference). However, being agnostic of the underlying energy landscape, the accuracy of such generative model may still be limited. In this work, we explore the few-shot setting of such pre-trained generative sampler which incorporates MD simulations in a tractable manner. Specifically, given a target protein of interest, we first acquire some seeding conformations from the pre-trained sampler followed by a number of physical simulations in parallel starting from these seeding samples. Then we fine-tuned the generative model using the simulation trajectories above to become a target-specific sampler. Experimental results demonstrated the superior performance of such few-shot conformation sampler at a tractable computational cost.
ProtIR: Iterative Refinement between Retrievers and Predictors for Protein Function Annotation
Feb 10, 2024
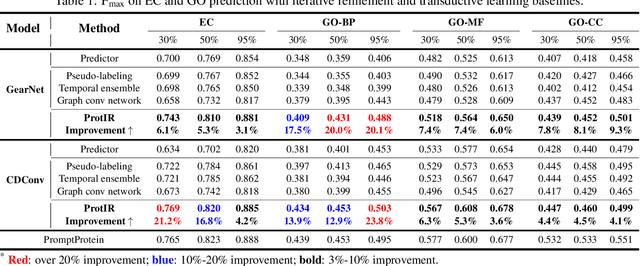
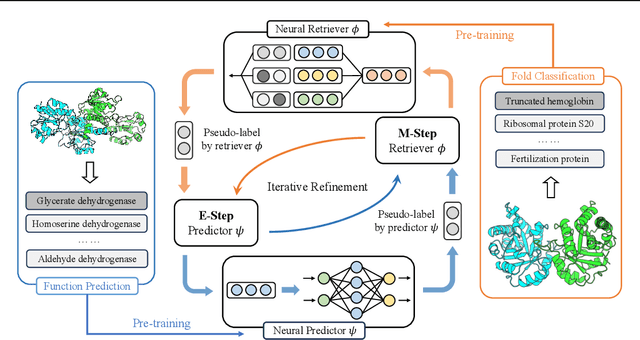

Protein function annotation is an important yet challenging task in biology. Recent deep learning advancements show significant potential for accurate function prediction by learning from protein sequences and structures. Nevertheless, these predictor-based methods often overlook the modeling of protein similarity, an idea commonly employed in traditional approaches using sequence or structure retrieval tools. To fill this gap, we first study the effect of inter-protein similarity modeling by benchmarking retriever-based methods against predictors on protein function annotation tasks. Our results show that retrievers can match or outperform predictors without large-scale pre-training. Building on these insights, we introduce a novel variational pseudo-likelihood framework, ProtIR, designed to improve function predictors by incorporating inter-protein similarity modeling. This framework iteratively refines knowledge between a function predictor and retriever, thereby combining the strengths of both predictors and retrievers. ProtIR showcases around 10% improvement over vanilla predictor-based methods. Besides, it achieves performance on par with protein language model-based methods, yet without the need for massive pre-training, highlighting the efficacy of our framework. Code will be released upon acceptance.
Structure-Informed Protein Language Model
Feb 07, 2024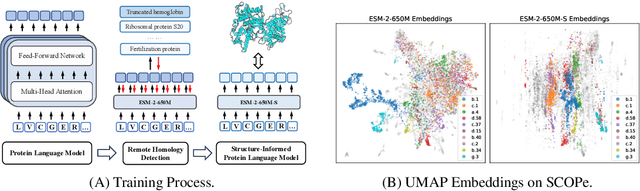

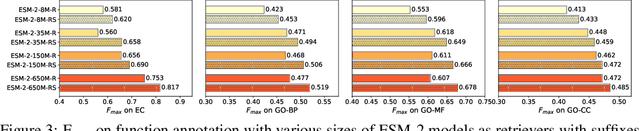
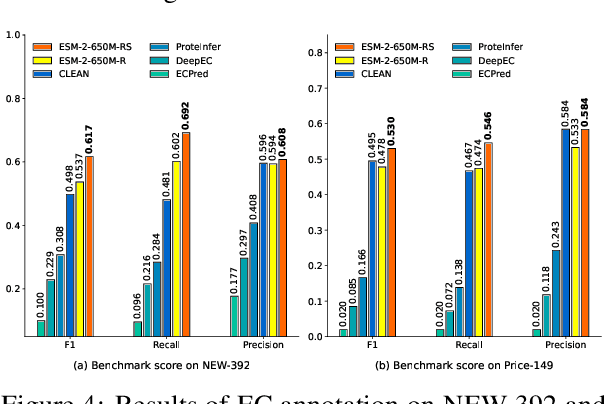
Protein language models are a powerful tool for learning protein representations through pre-training on vast protein sequence datasets. However, traditional protein language models lack explicit structural supervision, despite its relevance to protein function. To address this issue, we introduce the integration of remote homology detection to distill structural information into protein language models without requiring explicit protein structures as input. We evaluate the impact of this structure-informed training on downstream protein function prediction tasks. Experimental results reveal consistent improvements in function annotation accuracy for EC number and GO term prediction. Performance on mutant datasets, however, varies based on the relationship between targeted properties and protein structures. This underscores the importance of considering this relationship when applying structure-aware training to protein function prediction tasks. Code and model weights are available at https://github.com/DeepGraphLearning/esm-s.
PDB-Struct: A Comprehensive Benchmark for Structure-based Protein Design
Nov 30, 2023



Structure-based protein design has attracted increasing interest, with numerous methods being introduced in recent years. However, a universally accepted method for evaluation has not been established, since the wet-lab validation can be overly time-consuming for the development of new algorithms, and the $\textit{in silico}$ validation with recovery and perplexity metrics is efficient but may not precisely reflect true foldability. To address this gap, we introduce two novel metrics: refoldability-based metric, which leverages high-accuracy protein structure prediction models as a proxy for wet lab experiments, and stability-based metric, which assesses whether models can assign high likelihoods to experimentally stable proteins. We curate datasets from high-quality CATH protein data, high-throughput $\textit{de novo}$ designed proteins, and mega-scale experimental mutagenesis experiments, and in doing so, present the $\textbf{PDB-Struct}$ benchmark that evaluates both recent and previously uncompared protein design methods. Experimental results indicate that ByProt, ProteinMPNN, and ESM-IF perform exceptionally well on our benchmark, while ESM-Design and AF-Design fall short on the refoldability metric. We also show that while some methods exhibit high sequence recovery, they do not perform as well on our new benchmark. Our proposed benchmark paves the way for a fair and comprehensive evaluation of protein design methods in the future. Code is available at https://github.com/WANG-CR/PDB-Struct.
DiffPack: A Torsional Diffusion Model for Autoregressive Protein Side-Chain Packing
Jun 01, 2023Proteins play a critical role in carrying out biological functions, and their 3D structures are essential in determining their functions. Accurately predicting the conformation of protein side-chains given their backbones is important for applications in protein structure prediction, design and protein-protein interactions. Traditional methods are computationally intensive and have limited accuracy, while existing machine learning methods treat the problem as a regression task and overlook the restrictions imposed by the constant covalent bond lengths and angles. In this work, we present DiffPack, a torsional diffusion model that learns the joint distribution of side-chain torsional angles, the only degrees of freedom in side-chain packing, by diffusing and denoising on the torsional space. To avoid issues arising from simultaneous perturbation of all four torsional angles, we propose autoregressively generating the four torsional angles from \c{hi}1 to \c{hi}4 and training diffusion models for each torsional angle. We evaluate the method on several benchmarks for protein side-chain packing and show that our method achieves improvements of 11.9% and 13.5% in angle accuracy on CASP13 and CASP14, respectively, with a significantly smaller model size (60x fewer parameters). Additionally, we show the effectiveness of our method in enhancing side-chain predictions in the AlphaFold2 model. Code will be available upon the accept.
Enhancing Protein Language Models with Structure-based Encoder and Pre-training
Mar 11, 2023Protein language models (PLMs) pre-trained on large-scale protein sequence corpora have achieved impressive performance on various downstream protein understanding tasks. Despite the ability to implicitly capture inter-residue contact information, transformer-based PLMs cannot encode protein structures explicitly for better structure-aware protein representations. Besides, the power of pre-training on available protein structures has not been explored for improving these PLMs, though structures are important to determine functions. To tackle these limitations, in this work, we enhance the PLMs with structure-based encoder and pre-training. We first explore feasible model architectures to combine the advantages of a state-of-the-art PLM (i.e., ESM-1b1) and a state-of-the-art protein structure encoder (i.e., GearNet). We empirically verify the ESM-GearNet that connects two encoders in a series way as the most effective combination model. To further improve the effectiveness of ESM-GearNet, we pre-train it on massive unlabeled protein structures with contrastive learning, which aligns representations of co-occurring subsequences so as to capture their biological correlation. Extensive experiments on EC and GO protein function prediction benchmarks demonstrate the superiority of ESM-GearNet over previous PLMs and structure encoders, and clear performance gains are further achieved by structure-based pre-training upon ESM-GearNet. Our implementation is available at https://github.com/DeepGraphLearning/GearNet.
Physics-Inspired Protein Encoder Pre-Training via Siamese Sequence-Structure Diffusion Trajectory Prediction
Jan 28, 2023Pre-training methods on proteins are recently gaining interest, leveraging either protein sequences or structures, while modeling their joint energy landscape is largely unexplored. In this work, inspired by the success of denoising diffusion models, we propose the DiffPreT approach to pre-train a protein encoder by sequence-structure multimodal diffusion modeling. DiffPreT guides the encoder to recover the native protein sequences and structures from the perturbed ones along the multimodal diffusion trajectory, which acquires the joint distribution of sequences and structures. Considering the essential protein conformational variations, we enhance DiffPreT by a physics-inspired method called Siamese Diffusion Trajectory Prediction (SiamDiff) to capture the correlation between different conformers of a protein. SiamDiff attains this goal by maximizing the mutual information between representations of diffusion trajectories of structurally-correlated conformers. We study the effectiveness of DiffPreT and SiamDiff on both atom- and residue-level structure-based protein understanding tasks. Experimental results show that the performance of DiffPreT is consistently competitive on all tasks, and SiamDiff achieves new state-of-the-art performance, considering the mean ranks on all tasks. The source code will be released upon acceptance.
Metro: Memory-Enhanced Transformer for Retrosynthetic Planning via Reaction Tree
Sep 30, 2022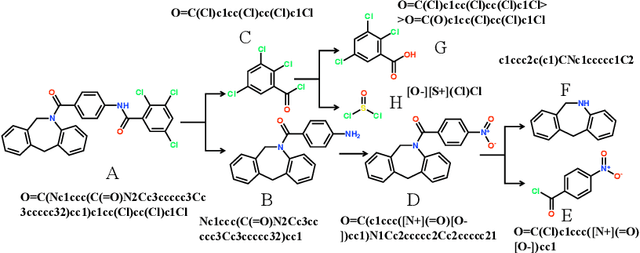
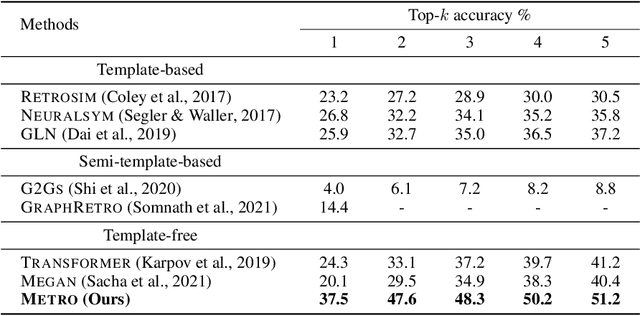
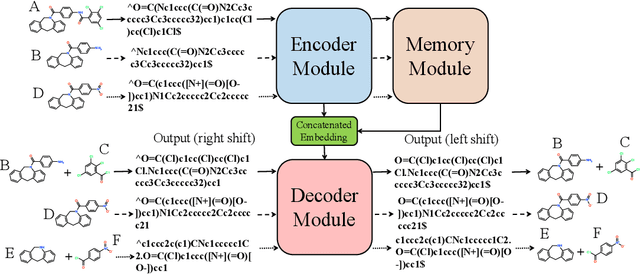
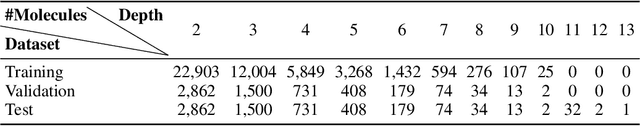
Retrosynthetic planning plays a critical role in drug discovery and organic chemistry. Starting from a target molecule as the root node, it aims to find a complete reaction tree subject to the constraint that all leaf nodes belong to a set of starting materials. The multi-step reactions are crucial because they determine the flow chart in the production of the Organic Chemical Industry. However, existing datasets lack curation of tree-structured multi-step reactions, and fail to provide such reaction trees, limiting models' understanding of organic molecule transformations. In this work, we first develop a benchmark curated for the retrosynthetic planning task, which consists of 124,869 reaction trees retrieved from the public USPTO-full dataset. On top of that, we propose Metro: Memory-Enhanced Transformer for RetrOsynthetic planning. Specifically, the dependency among molecules in the reaction tree is captured as context information for multi-step retrosynthesis predictions through transformers with a memory module. Extensive experiments show that Metro dramatically outperforms existing single-step retrosynthesis models by at least 10.7% in top-1 accuracy. The experiments demonstrate the superiority of exploiting context information in the retrosynthetic planning task. Moreover, the proposed model can be directly used for synthetic accessibility analysis, as it is trained on reaction trees with the shortest depths. Our work is the first step towards a brand new formulation for retrosynthetic planning in the aspects of data construction, model design, and evaluation. Code is available at https://github.com/SongtaoLiu0823/metro.